





TMT-Based Quantitative Proteomics: Multiplexing, MS3 Accuracy, and Workflow Design
- TMT supports multiplexed analysis by labeling peptides from different samples with isobaric tags that separate at the reporter-ion level.
- Reporter-ion quantification is usually extracted from MS2 or MS3 spectra, with MS3 commonly improving quantitative accuracy in interference-prone samples.
- Pooling labeled samples reduces technical variability across runs and improves consistency for comparative studies.
- High-pH fractionation, balanced experimental design, and interference control are central to deep and reliable TMT workflows.
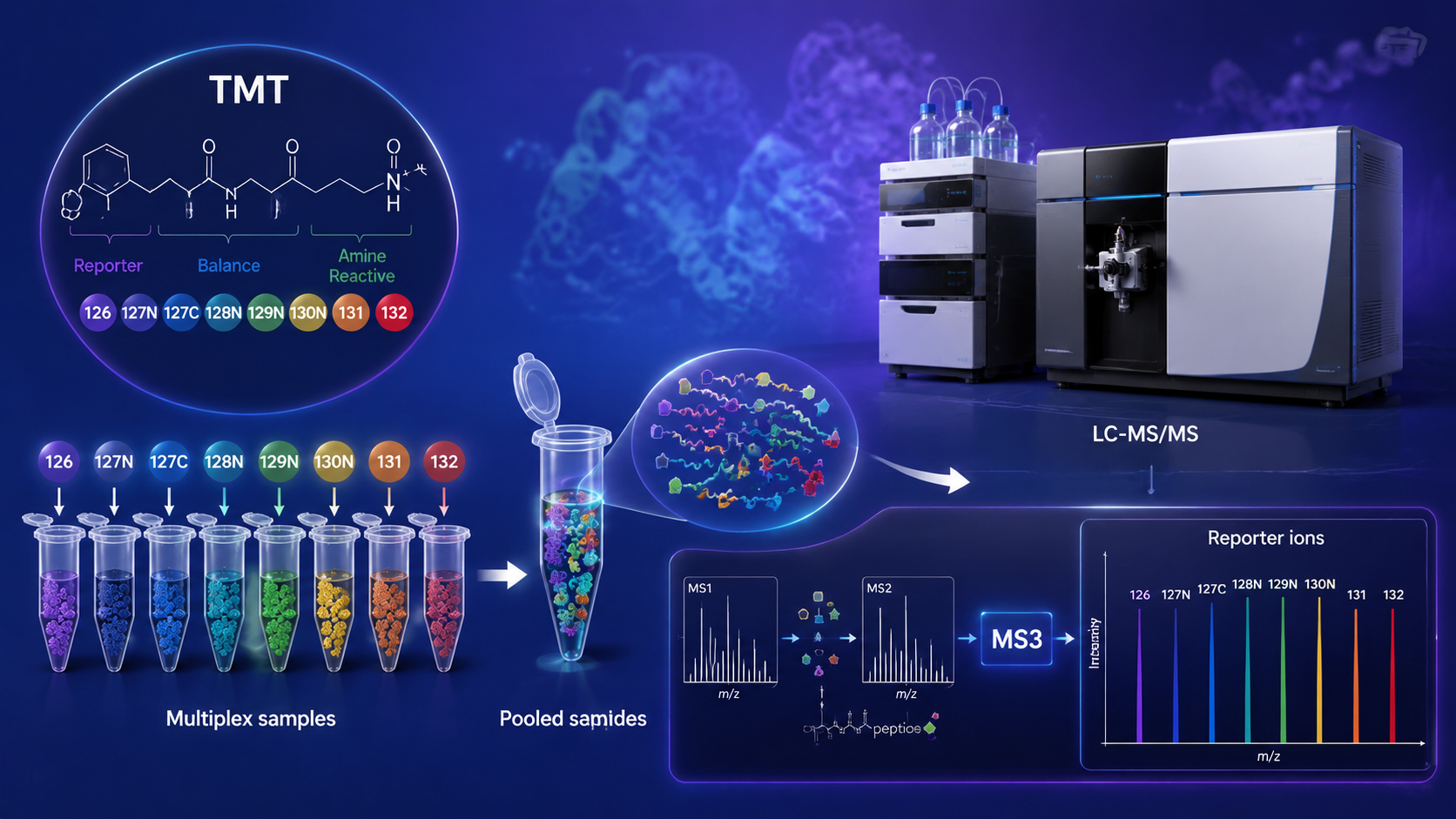
TMT-based quantitative proteomics is an isobaric labeling workflow in which peptides from different biological samples are tagged with tandem mass tags, pooled, fractionated, and quantified through reporter ions generated during tandem mass spectrometry. The method is widely used when researchers need high-throughput, low-variation comparison across many samples, such as clinical cohorts, treatment-response experiments, biomarker studies, and time-course designs.
Key Takeaways
What TMT Does in Proteomics?
TMT reagents are designed so labeled peptides remain isobaric at the precursor level but release channel-specific reporter ions after fragmentation. That allows many biological samples to be measured together in one experiment.
Related Services
TMT Quantitative Proteomics Analysis Service
Tandem Mass Tag (TMT) Technology Service
iTRAQ/TMT/MultiNotch Quantitative Proteomics Service
TMT/iTRAQ Labeling-Based Quantitative Service
Standard TMT Workflow
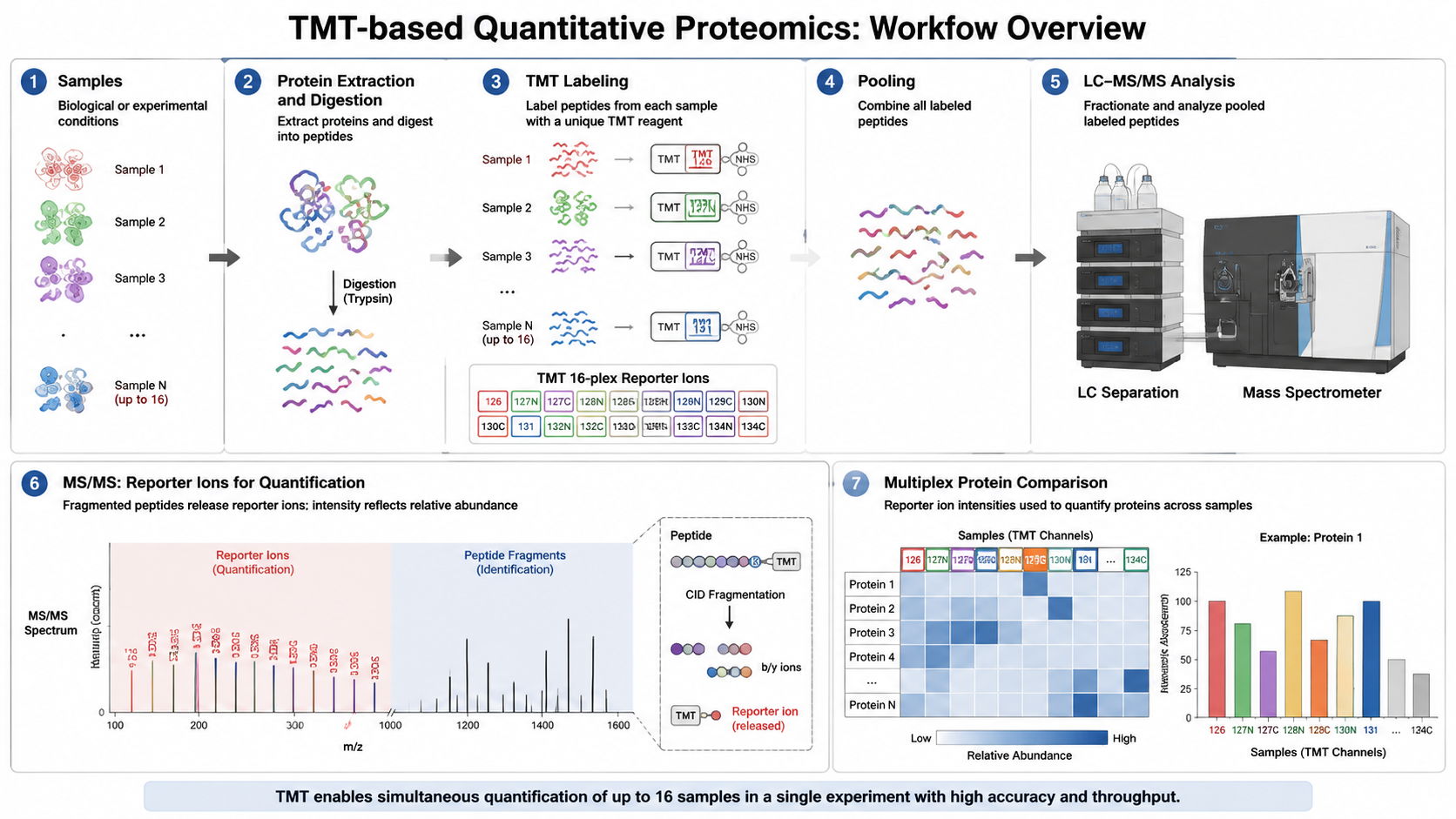
A typical TMT experiment includes protein extraction, digestion, peptide cleanup, channel-specific TMT labeling, equal mixing, peptide fractionation, nanoLC-MS/MS, peptide identification, reporter-ion extraction, normalization, and downstream differential protein analysis.
Balanced channel assignment matters. If all controls sit in adjacent channels or if batches are not bridged correctly, downstream normalization becomes harder.
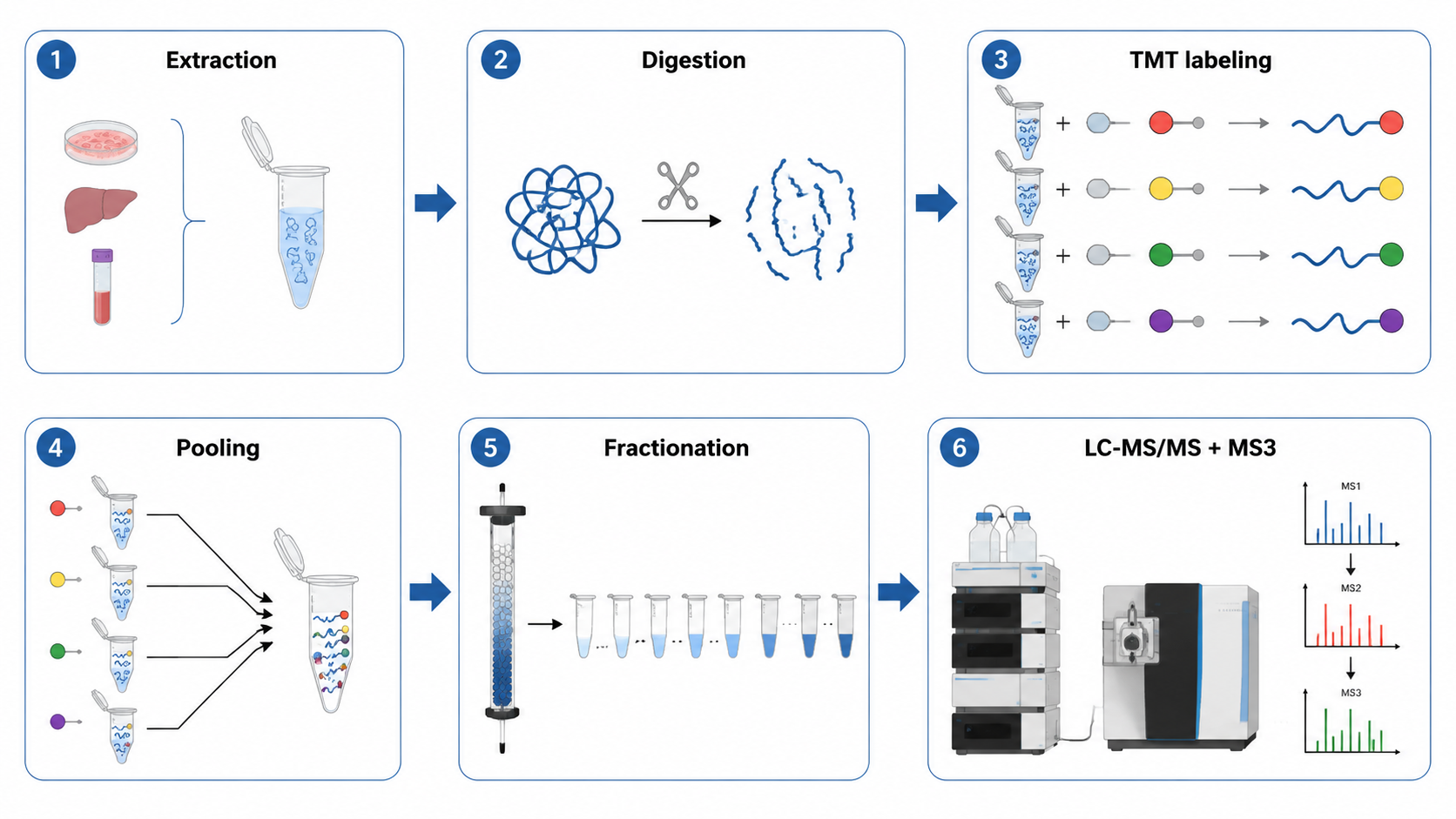
Why MS3 is often Discussed in TMT Workflows?
Co-isolation interference can distort reporter-ion intensities when unrelated peptide fragments contribute signal in the same MS2 spectrum. MultiNotch SPS-MS3 workflows reduce that problem by re-fragmenting selected MS2 ions before quantification.
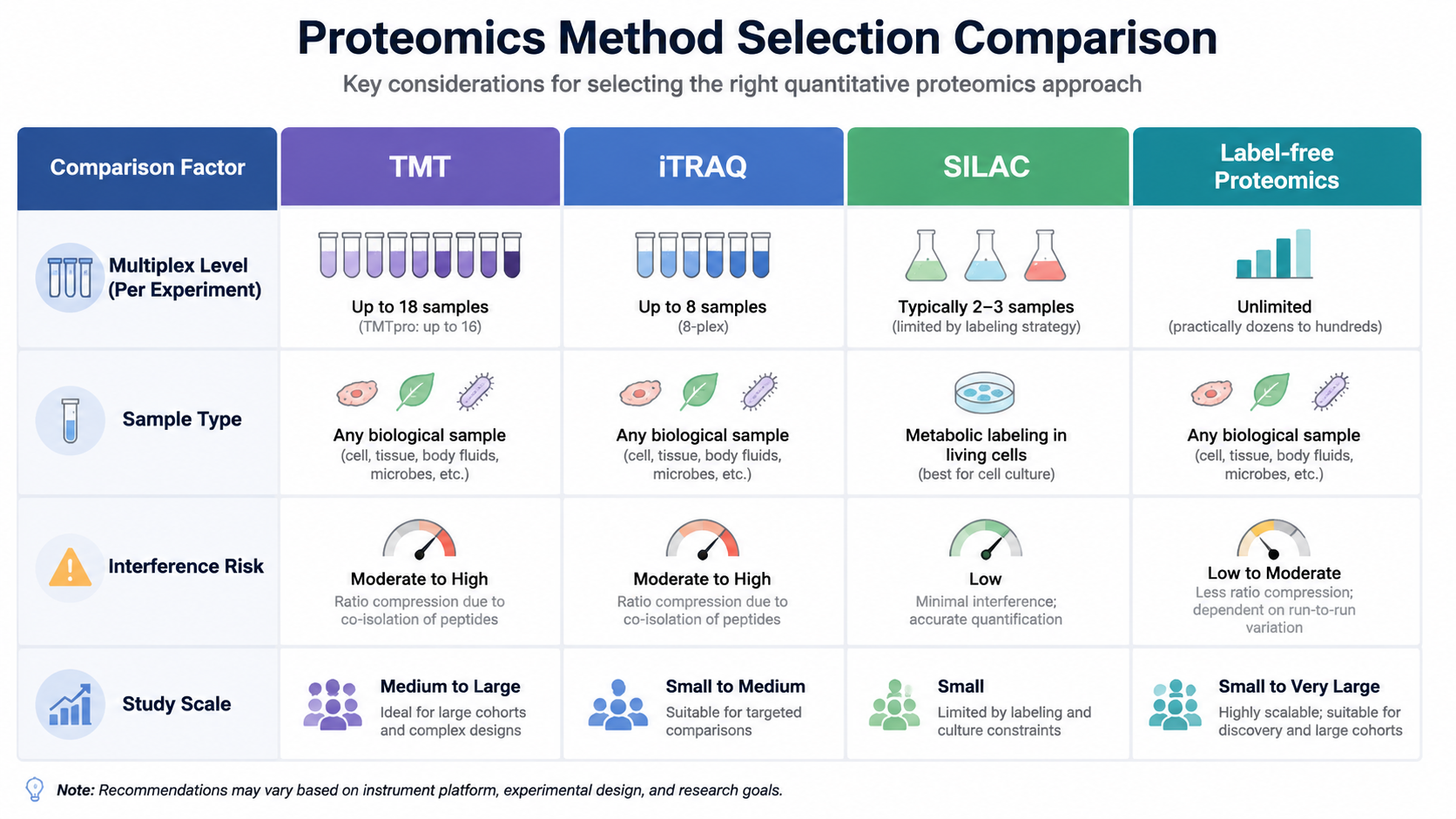
TMT vs Label-Free and other Label-Based Methods
| Strategy | Main strength | Main caution | Best fit |
|---|---|---|---|
| TMT | High multiplexing and cross-sample consistency | Interference and workflow complexity | Large comparative cohorts |
| iTRAQ | Established multiplex chemistry | Lower plex and similar interference concerns | Moderate multiplex comparative studies |
| SILAC | Clean metabolic labeling | Mostly limited to cell culture | Controlled cellular systems |
| Label-free | Simple chemistry-free workflow | Higher run-to-run dependence | Flexible discovery studies |
Common Applications
TMT is widely used for disease mechanism studies, drug response profiling, biomarker discovery, tissue comparison, time-course proteomics, and multi-omics integration.
Practical Design Tips
Use pooled references or bridge channels for multi-batch studies. Keep sample handling consistent before labeling. Fractionate enough to reduce interference without generating unmanageable data volume.
FAQ
1. What is TMT-based quantitative proteomics?
It is a mass-spectrometry workflow that labels peptides from different samples with tandem mass tags, then quantifies relative abundance through reporter ions released during fragmentation.
2. Why use TMT instead of label-free proteomics?
TMT reduces inter-run variation by pooling labeled samples and supports multiplex comparison across many conditions or time points.
3. What is the benefit of MS3 in TMT?
MS3 can reduce ratio distortion caused by co-isolated precursor interference, improving quantitative confidence in complex samples.
Conclusion
TMT-based quantitative proteomics is a powerful workflow for multiplex comparative biology when researchers need high throughput, strong cross-sample consistency, and structured cohort analysis. The most reliable studies combine balanced experimental design, efficient labeling, strong fractionation, interference-aware MS acquisition, and biological validation of the most important quantitative findings.
How to order?

