





DIA vs DDA Proteomics: Acquisition Strategy, Quantification, and Method Selection
- DDA is intensity-driven: the instrument selects the top precursor ions for MS/MS.
- DIA is window-driven: the instrument fragments all precursors within repeated m/z isolation windows.
- DDA produces cleaner MS/MS spectra but can miss low-abundance peptides across runs.
- DIA improves reproducibility and quantification but produces more complex spectra.
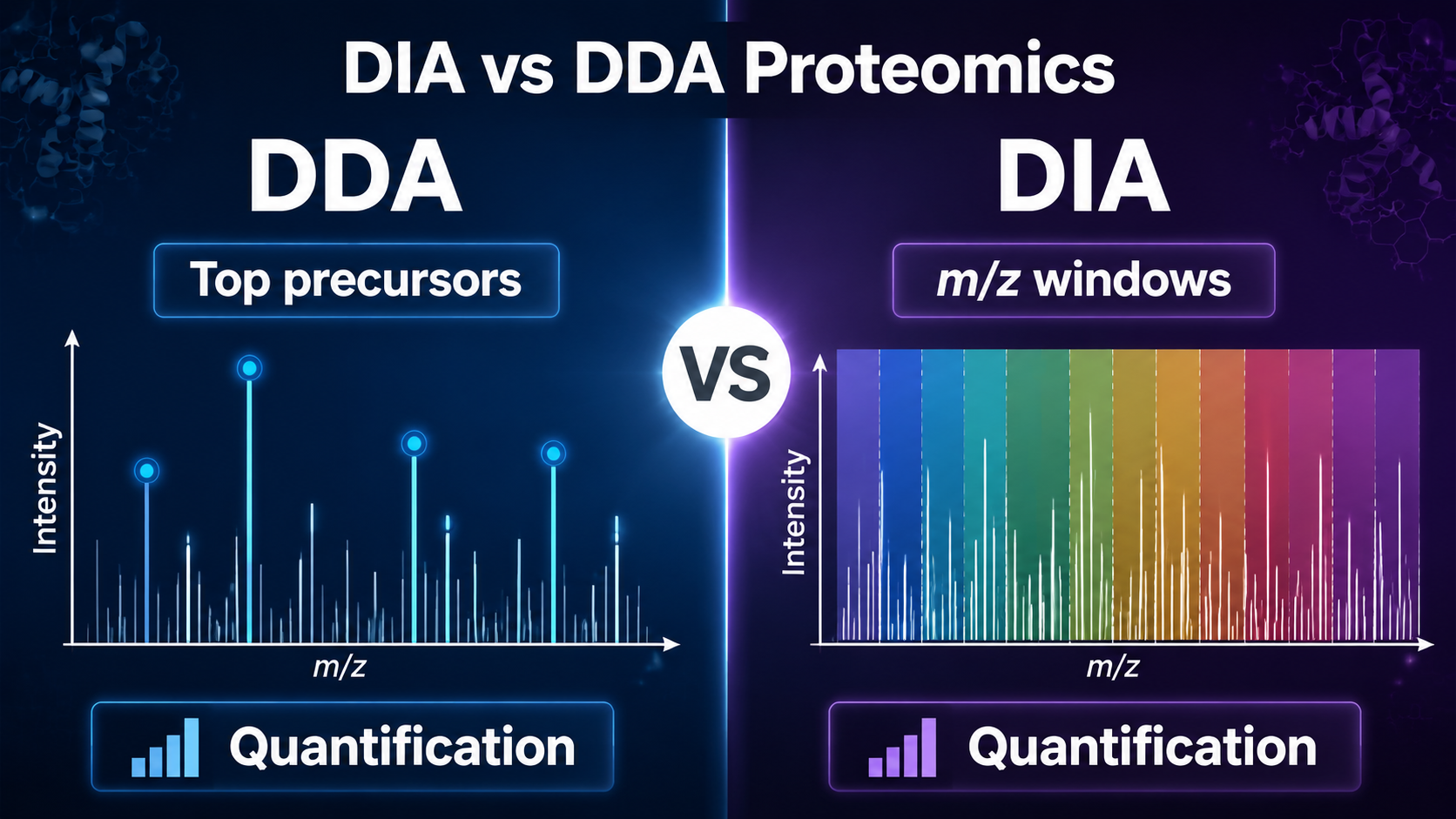
DDA and DIA are two LC-MS/MS acquisition strategies used in proteomics. DDA selects the most intense precursor ions in real time for fragmentation, while DIA fragments all detectable ions across predefined m/z windows. In practice, DDA is often preferred for discovery, spectral library generation, and high-confidence spectra, whereas DIA is preferred for reproducible quantification, large cohorts, low missing values, and biomarker studies.
Key Takeaways
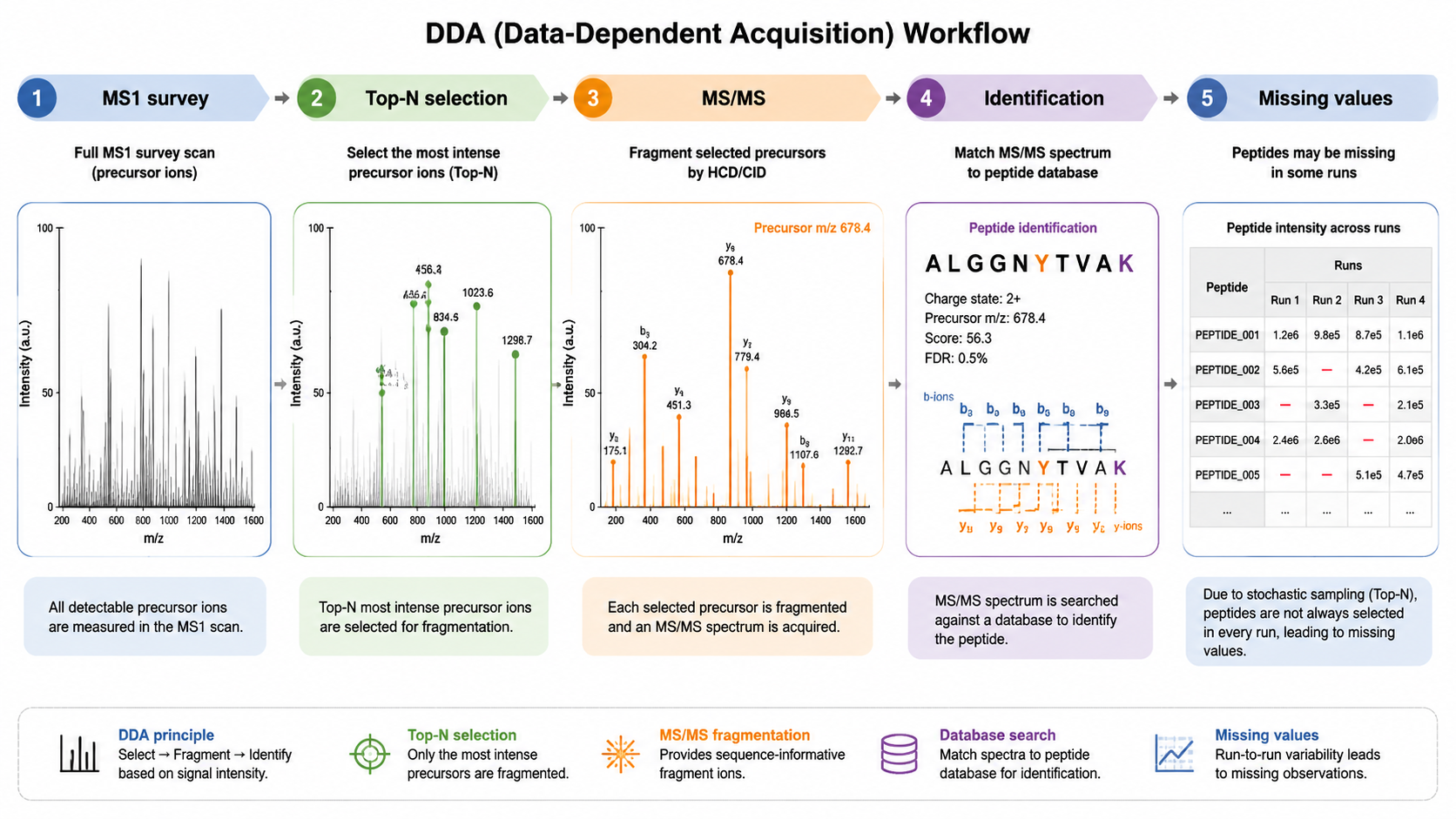
How DDA Works?
Data-dependent acquisition starts with an MS1 survey scan. The instrument then selects the top N most intense precursor ions for MS/MS fragmentation. This approach often produces high-quality spectra for peptide identification because each MS/MS spectrum contains fewer cofragmented precursors. The weakness is stochastic sampling: a peptide detected in one run may not be selected in another.
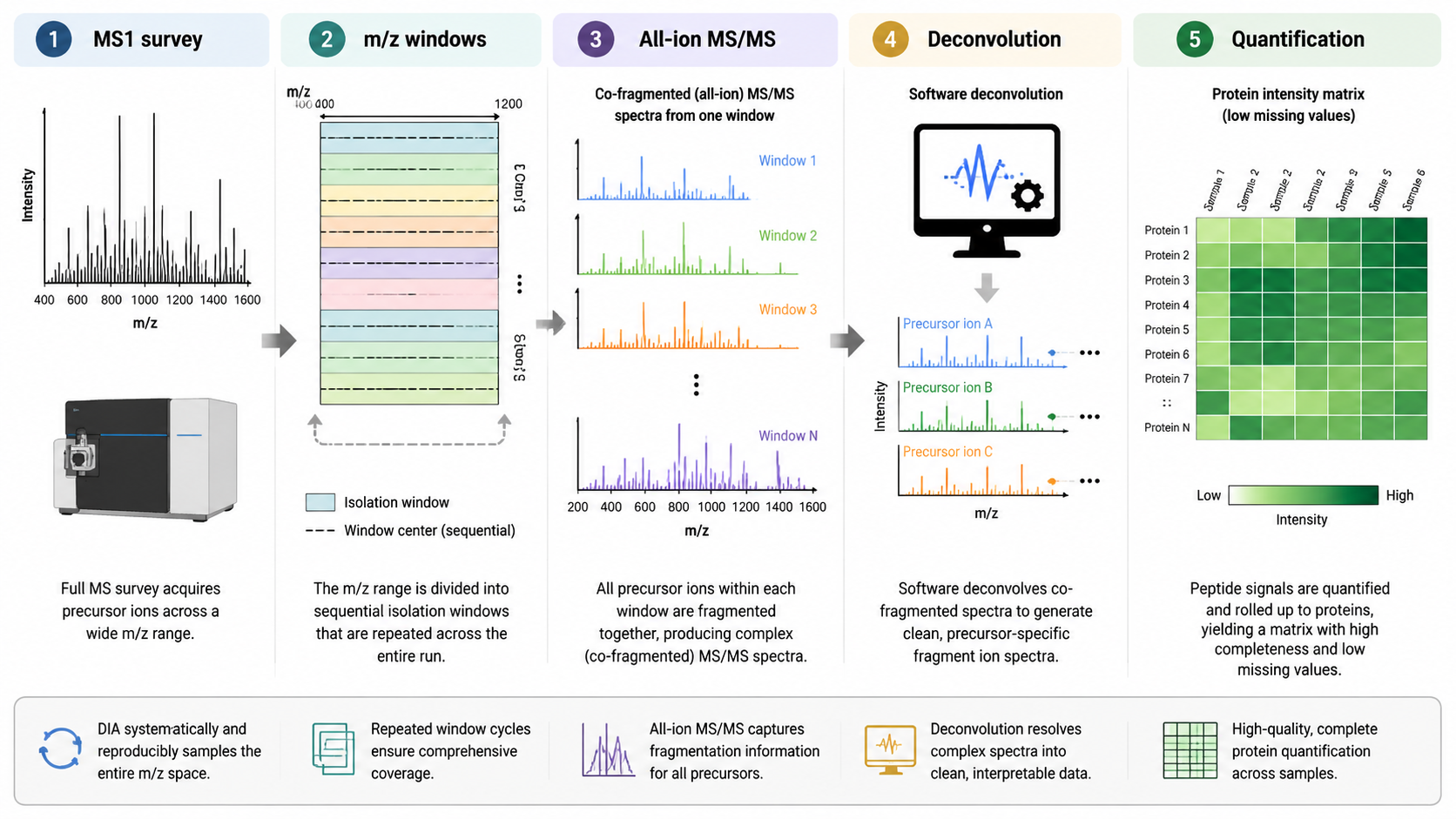
How DIA Works?
Data-independent acquisition divides the m/z range into sequential windows and fragments all precursors within each window. DIA reduces selection bias and improves consistency across samples, but each MS/MS spectrum contains fragments from multiple precursor ions. DIA data analysis therefore relies heavily on algorithms, spectral libraries, predicted spectra, or library-free search methods.
Related Services
DIA Proteomics Analysis Service
4D-DIA Quantitative Proteomics Service
DIA vs DDA Comparison
| Feature | DDA | DIA | Practical meaning |
|---|---|---|---|
| Precursor selection | Top-intensity ions | All ions in m/z windows | DIA is less dependent on real-time abundance ranking |
| MS/MS spectra | Cleaner, fewer mixed precursors | More complex, cofragmented | DDA is easier to interpret manually |
| Reproducibility | Lower across many runs | Higher across many runs | DIA is better for cohorts |
| Missing values | More common | Reduced | DIA improves statistical power |
| Best use | Discovery and library generation | Quantification and biomarker studies | Many projects use both |
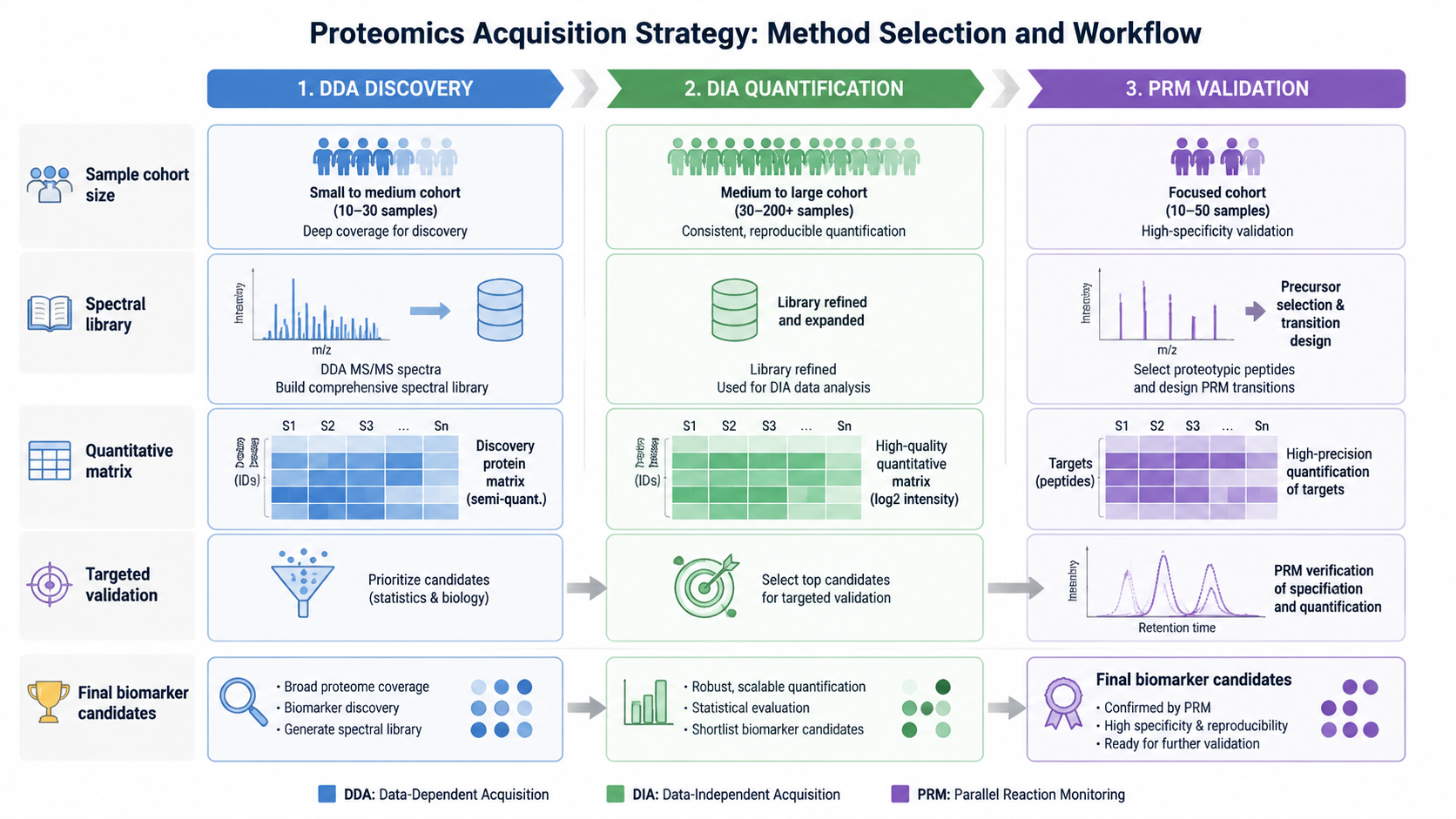
When to Choose DDA?
DDA is useful when the goal is initial protein discovery, high-confidence MS/MS interpretation, spectral library construction, or studies where clean spectra matter. It is also suitable for building a reference dataset before downstream DIA quantification.
When to Choose DIA?
DIA is useful when reproducible quantification is the priority. It is commonly selected for clinical cohorts, differential protein abundance studies, biomarker discovery, low-abundance protein detection, and projects where missing values would weaken statistics.
FAQ
1. What is the main difference between DDA and DIA?
DDA selects a subset of high-intensity precursor ions for fragmentation. DIA fragments all ions within predefined m/z windows, creating more comprehensive but more complex MS/MS data.
2. Is DIA better than DDA?
DIA is often better for reproducible quantification and large sample sets. DDA can be better for clean spectra, discovery, and spectral library generation. The better method depends on the study goal.
3. Why does DDA have more missing values?
DDA selects precursors based on real-time signal intensity. Low-abundance peptides may be selected in one run but skipped in another, causing missing values.
4. Can DDA and DIA be combined?
Yes. A common strategy is to use DDA to build spectral libraries, DIA for large-scale quantification, and PRM or MRM for targeted validation.
Conclusion
DDA and DIA are not competing labels as much as different acquisition choices. DDA gives cleaner spectra and strong discovery utility. DIA gives more consistent quantification and fewer missing values. The strongest proteomics designs often combine them: DDA for library or discovery, DIA for cohort quantification, and targeted MS for validation.
How to order?

