





Why Antibody Sequencing Is Difficult: Key Technical Barriers in Real-World Sequence Recovery
- Best-case input: purified monoclonal antibody, simple buffer, intact chains, enough material for multi-enzyme protease digestion
- Most common barriers: isoleucine/leucine ambiguity, incomplete peptide coverage in variable regions, post-translational modification (PTM) heterogeneity, mixed composition, and no DNA or hybridoma for orthogonal validation
- Likely deliverable when barriers remain: confidence-ranked heavy-chain and light-chain sequence assembly, not a fully closed sequence
- Core feasibility question: can the current sample support residue-level confidence in the variable region, or only a validation-dependent reconstruction?
- heavy-chain and light-chain peptide maps
- sequence tags and assembled candidate sequences
- chain assignment with stated confidence
- location of ambiguous residues
- identification of PTM-bearing peptides that affect interpretation
Antibody de novo sequencing becomes hard when a project needs clone-relevant residues from incomplete or ambiguous LC-MS/MS evidence, not just a match to a known database entry. In practice, interpretable sequence recovery is most likely when the sample produces clean, overlapping peptides across both the heavy chain and light chain, especially in the variable region and the complementarity-determining region (CDR). When the sample is mixed, rich in modifications, obscured by formulation background, or missing peptide overlap in key CDR-heavy segments, the practical result is often partial sequence recovery with unresolved positions.
Quick decision block
The first practical split is simple: is the main limit coming from sample composition, or from spectrum-to-sequence interpretation? In real projects, the biggest problems usually are not in the framework region. They tend to appear as sequence gaps in variable domains, uncertain residue calls, modification-affected spectra, and uncertainty about whether the recovered peptides actually come from one antibody clone.
Where antibody sequencing usually breaks down
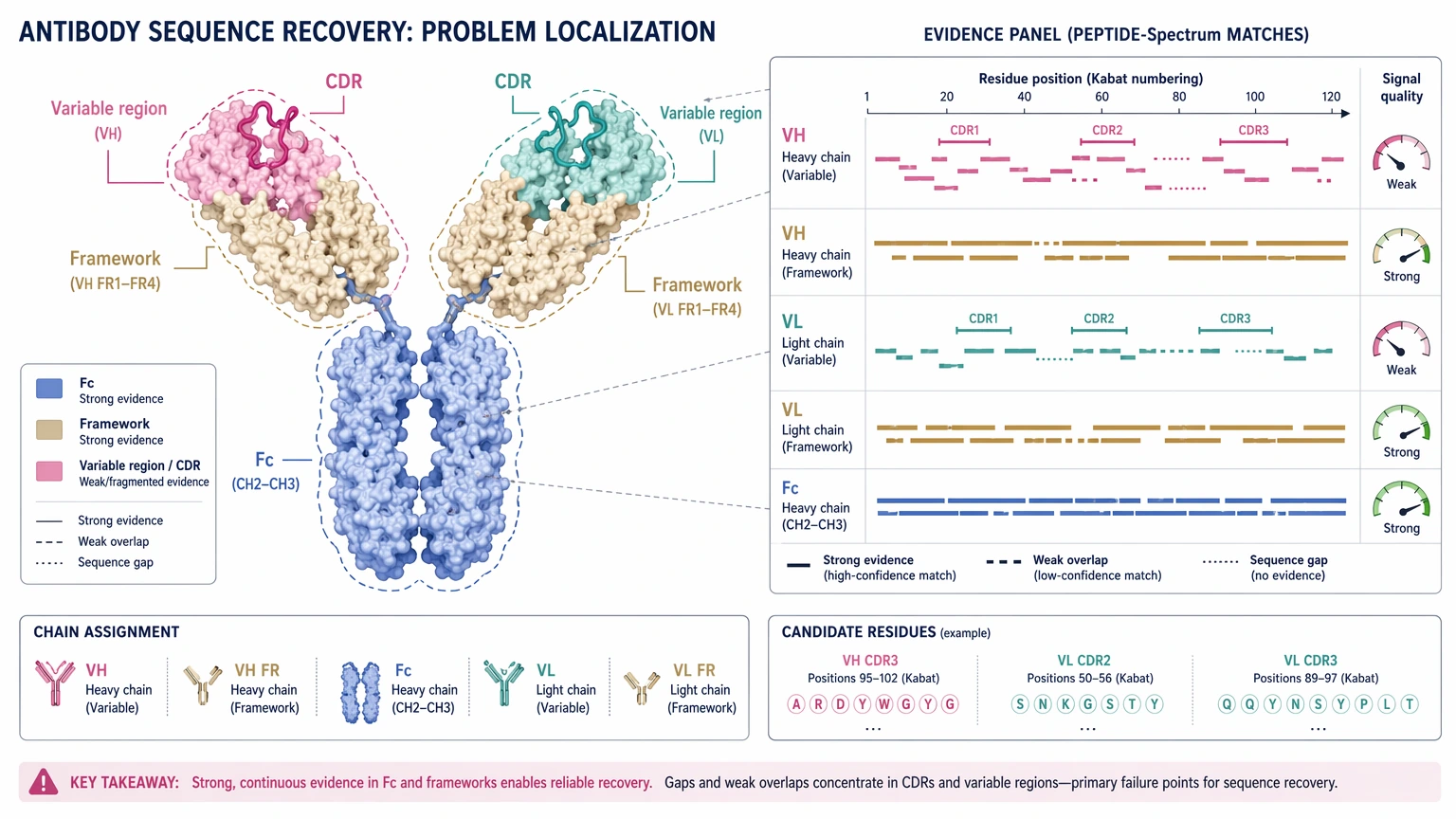
Many teams assume that a purified antibody is automatically ready for sequencing. That often turns out to be wrong. The recovered data may show strong Fc or framework-region evidence but weak sequence continuity in the variable region. A report may include peptide mapping results and still leave chain assignment uncertain, peptide overlap discontinuous, or several candidate residues clustered in one CDR segment.
This comes up most often in three situations. First, the original DNA, hybridoma, or expression construct is gone, so LC-MS/MS has to carry most of the sequence burden. Second, an earlier bottom-up sequencing attempt delivered only partial heavy-chain or light-chain recovery. Third, material labeled as purified antibody still contains carrier proteins, clipped species, stabilizers, or a low-level mixed antibody population.
That distinction matters because the project goal is rarely just “get any sequence.” More often, the real goal is to reconstruct the variable region, confirm clone-specific residues, or decide whether partial recovery is enough for engineering or comparability work. In other words, antibody de novo sequencing is not mainly about total peptide count. It is about whether the evidence is strong enough to assemble the parts of the antibody that actually distinguish the clone.
The most relevant technical barriers
Variable-region peptides are harder to recover than conserved regions
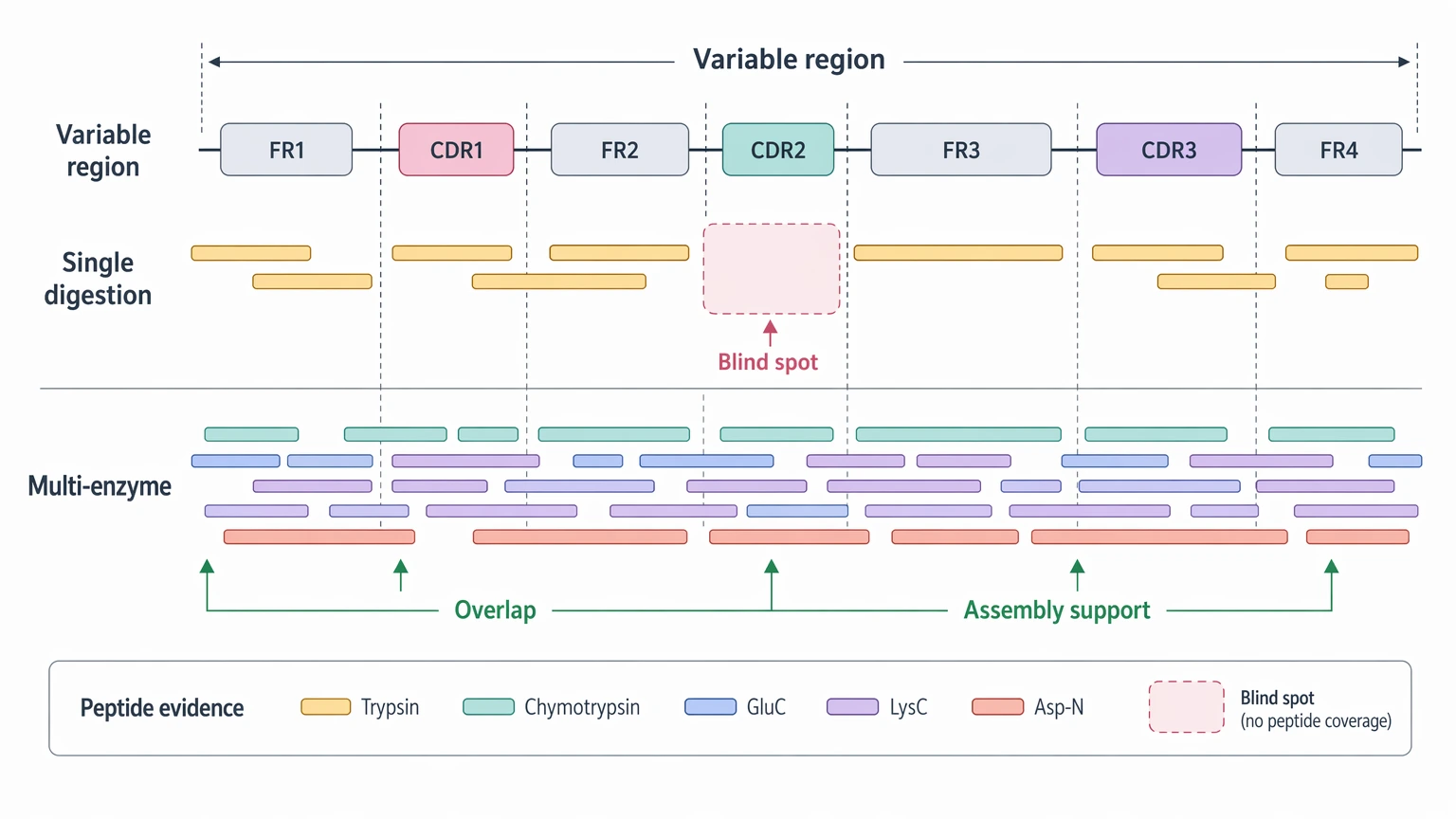
Conserved regions often yield tractable peptides and recognizable sequence tags. The residues that matter most, though, are usually concentrated in the variable region, especially in the CDR. Those peptides may be short, fragment poorly, or simply not appear under a given protease digestion pattern. So even when total peptide coverage looks acceptable, the evidence may still be thin exactly where clone identity is defined.
This is one reason standard protein identification logic does not transfer neatly to unknown antibodies. For a known protein, partial matching may still be enough for identification. For antibody de novo sequencing, partial matching can leave the most important residues unresolved.
Isoleucine/leucine ambiguity is a real residue-level limit
Standard tandem mass spectrometry usually cannot distinguish isoleucine from leucine because they are isobaric. In routine protein identification, that may be a minor inconvenience. In antibody sequence recovery, it can be the deciding issue if the ambiguous position sits in a CDR or another clone-defining segment.
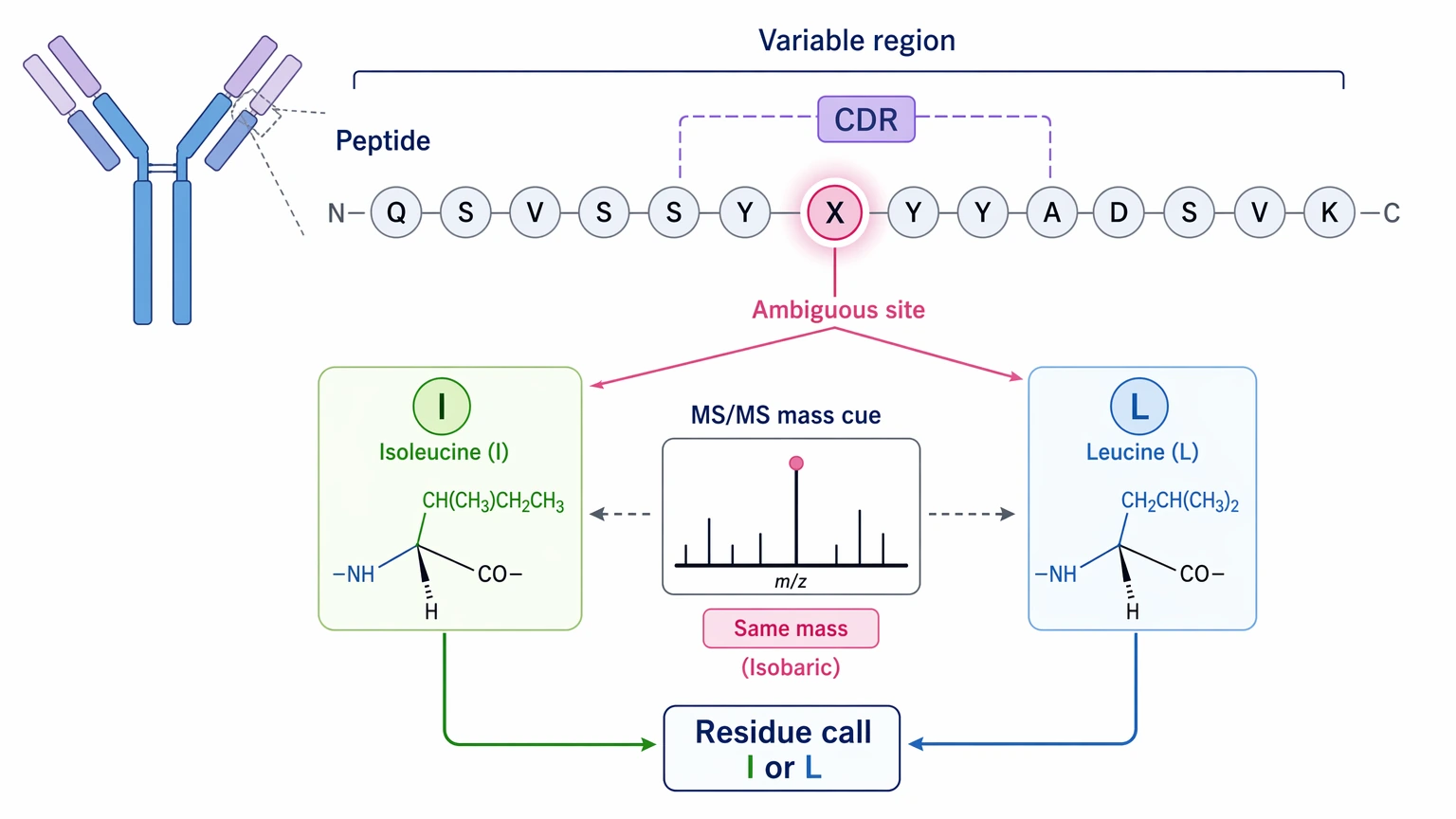
So a sequence can look structurally plausible and still remain uncertain at specific positions. One limit should be stated plainly here: bottom-up LC-MS/MS alone cannot be assumed to resolve every isoleucine/leucine position, even when overall peptide evidence is strong. Follow-up confirmation may still be necessary.
PTMs and glycosylation complicate MS/MS interpretation
Antibodies are not single uniform molecules. They often contain glycosylation, oxidation, deamidation, pyroglutamate formation, clipping, and other PTMs. Disulfide bond architecture also affects digestion behavior and peptide observability. Together, these features increase precursor heterogeneity and can split one expected peptide into several related forms.
For sequence interpretation, that does more than add complexity. Modified peptides may fragment less cleanly, compete with unmodified forms, or generate spectra that are harder to assign with high residue-level confidence. Database-search support is also limited when the target antibody is unknown, engineered, proprietary, or absent from reference libraries.
Mixed composition can undermine clone-specific recovery
A sample can pass routine purity checks and still be poor input for sequence reconstruction. Common interferences include bovine serum albumin, serum proteins, formulation excipients, clipped fragments, aggregates, or more than one antibody-derived species. Once mixed evidence enters the workflow, chain assignment becomes less certain, and sequence assembly may stop representing a single clone.
At that point, the question changes. It is no longer only “what residues are missing?” It becomes “does this peptide evidence belong to one antibody population at all?”
Missing source material removes a major confirmation route
When nucleic acid, hybridoma cells, or the original construct is available, uncertain residues can sometimes be checked outside the LC-MS/MS dataset. When none of that source material exists, de novo interpretation loses a major external anchor. That makes overlapping peptides, digestion design, and validation planning much more important before the sequencing project starts.
A troubleshooting path before you outsource
This subject works better as a troubleshooting workflow than as a generic step-by-step protocol. The goal is to identify the main blocker, narrow the expected deliverable, and decide whether the sample can support full antibody de novo sequencing or only ambiguity-managed recovery.
Step 1: Define the failure mode precisely
Separate low total peptide coverage, poor variable-region recovery, unresolved residues, and uncertain clone specificity. Those are different failure modes. A sample with good overall peptide mapping but weak CDR continuity needs a different response from a sample with broad contamination or severe degradation.
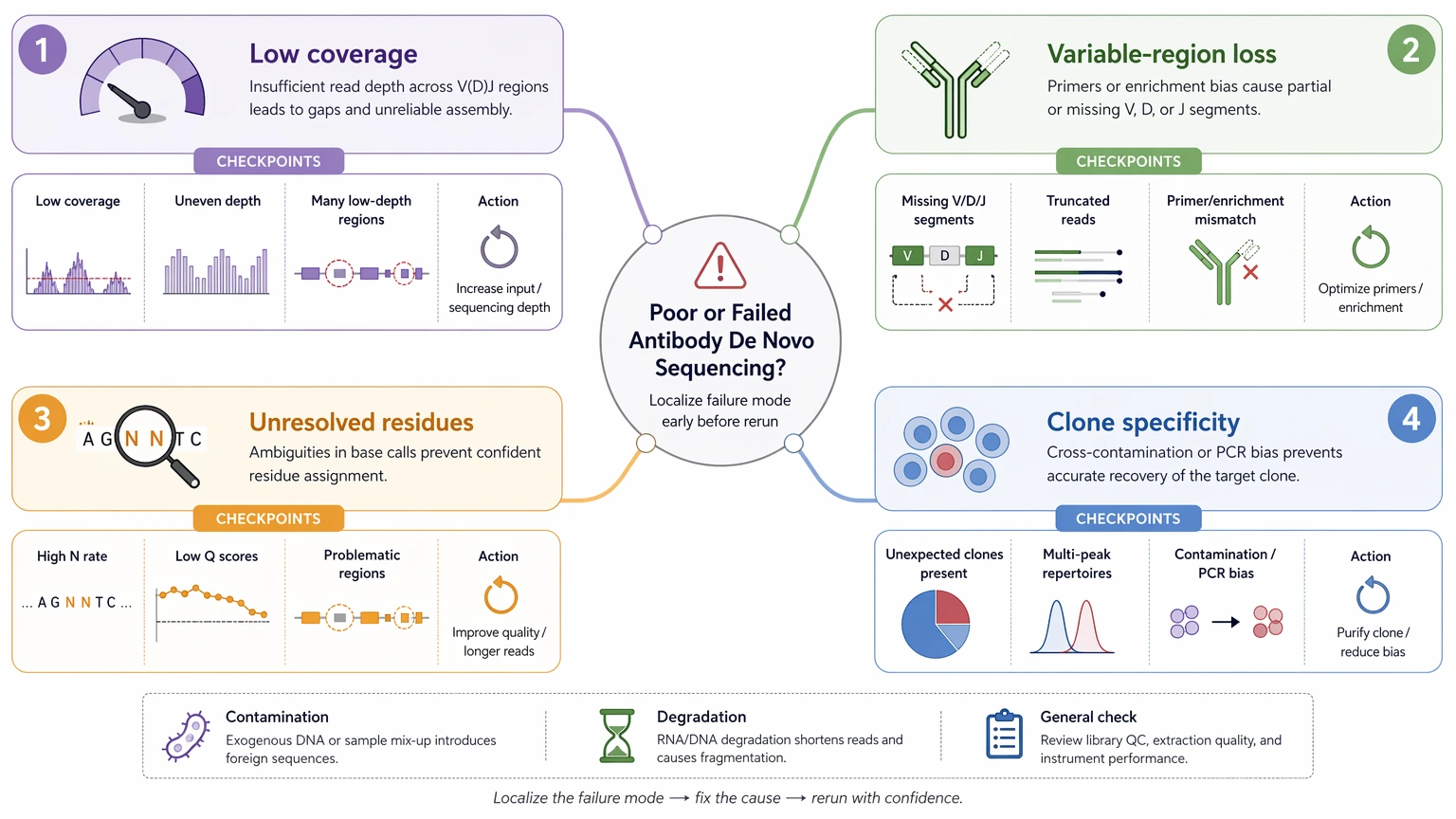
Step 2: Check sample composition before repeating LC-MS/MS
Review whether the input is an intact monoclonal antibody, a fragment mixture, a stored formulation sample, or a preparation with known additives. This early check often saves time because repeating the workflow does not fix a fundamentally mixed sample.
The table below can help frame the decision.
| Sample type | Best fit | Main limitation | Best next step |
|---|---|---|---|
| Purified monoclonal antibody in simple buffer | Broad feasibility assessment for bottom-up sequencing | Variable-region gaps may still remain | Use multi-enzyme peptide mapping |
| Antibody with excipients or carrier proteins | Pre-sequencing composition review | Background can interfere with chain assignment | Clean up sample and confirm composition |
| Degraded antibody or fragments | Partial region recovery | Domain continuity may be lost | Focus on recoverable regions and validation |
| Mixed antibody population | Limited clone-specific interpretation | Peptides may not map to one clone | Resolve composition before sequence assembly |
| PTM-heavy sample | Sequence-plus-PTM review | Modified peptides may lower confidence | Plan targeted follow-up for uncertain regions |
Takeaway: the sample label alone does not tell you how recoverable the sequence will be.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
Step 3: Ask whether peptide overlap exists where it matters
For antibody de novo sequencing, overlapping peptides in the variable region matter more than global coverage percentages. A single protease digestion can leave blind spots. Multi-enzyme protease digestion can improve overlap and support more stable sequence assembly, especially across CDR-adjacent regions.
Step 4: Decide how much ambiguity the project can tolerate
Some projects can move forward with a candidate sequence set plus clearly marked uncertain positions. Others need higher confidence because a small number of residues will affect engineering, intellectual property review, or clone reconstruction. If that tolerance is defined early, the workflow can match the real decision instead of overselling full closure.
This is often the point where a practical project discussion starts. If a prior vendor report already shows CDR gaps, PTM-heavy peptides, or uncertain chain assignment, you can submit your requirements and data context to MtoZ Biolabs to evaluate whether the next step should be additional digestion design, targeted validation, or a narrower recovery plan rather than another unchanged sequencing round.
Expected results and validation methods
A successful project does not always end with one fully unambiguous sequence. In antibody work, a technically meaningful result may be a ranked sequence assembly with strong framework support, substantial variable-region recovery, and a short list of unresolved positions.
Immediate deliverables from antibody de novo sequencing commonly include:
Follow-up confirmation is a different step. It focuses on whether the uncertain positions can be resolved well enough for the project goal. That may involve targeted LC-MS/MS review of specific peptides, orthogonal validation against source material if available, or focused confirmation of residues in a CDR or another functionally sensitive segment.
The key interpretation rule is straightforward: sequence recovery and biological equivalence are not the same claim. A partially recovered variable region can still be useful for clone tracking, rescue planning, or feasibility assessment, but it should not be treated as complete proof of exact clone identity.
Key cautions and practical limits
Several limits should be stated clearly before sequencing begins.
Sample quality or amount limits: low-input, degraded, or repeatedly freeze-thawed material may still produce data, but often with reduced peptide coverage and weaker overlap.
Controls and repeat expectations: one run may not answer the feasibility question. Replicate digestion or targeted follow-up may be necessary when critical residues remain uncertain.
Batch and contamination risk: formulation additives, background proteins, or low-level mixed species can distort chain assignment and create misleading sequence paths.
Interpretation boundaries: MS/MS evidence can support strong sequence inference, but residue-level confidence varies with peptide quality, PTM state, and overlap. Isoleucine/leucine ambiguity may stay unresolved without orthogonal evidence.
When another method or outside support is the better next step: if the sample is clearly mixed, heavily degraded, or missing decisive overlap in clone-defining regions, the better move may be cleanup, alternative confirmation, or a scoped feasibility review rather than direct full-sequence reconstruction. If you need that judgment before committing more sample, contact MtoZ Biolabs to evaluate the project context and discuss what output is technically realistic.
Conclusion
Antibody de novo sequencing is difficult because the residues that matter most often sit in variable-region peptides that are sparse, modification-affected, or intrinsically ambiguous in LC-MS/MS data. In practice, the strongest predictors of success are clean sample composition, recoverable heavy-chain and light-chain peptides, continuous overlapping peptides across CDR-relevant regions, and a realistic plan for orthogonal validation when residue-level confidence stays limited. For projects involving purified antibodies, failed vendor reports, missing genetic source material, or ambiguity-sensitive reconstruction goals, the most useful next step is a feasibility-focused review that defines what can be recovered now, what still needs confirmation, and whether the current sample supports the decision you need to make. If you are preparing that discussion, submit your requirements and sample context so the project scope can be assessed before more material is consumed.
FAQ
Can intact-mass data replace bottom-up sequencing for antibody recovery?
Usually no. Intact-mass analysis is useful for composition and heterogeneity assessment, but by itself it does not provide the peptide-level evidence needed for sequence assembly in variable regions.
Does heavy-chain sequencing usually fail more often than light-chain sequencing?
Not always, but heavy-chain interpretation is often harder because glycosylation and broader proteoform heterogeneity can complicate precursor selection and spectrum clarity.
If a vendor reports several candidate residues at one site, should that be treated as a failed project?
Not automatically. The result may still be technically useful if the uncertainty is localized, clearly reported, and outside the residues that drive the project decision.
Are recombinant or engineered antibodies harder to interpret than standard monoclonal antibodies?
They can be. Engineered regions, unusual PTMs, or noncanonical sequence features may reduce database support and make de novo interpretation more dependent on high-quality overlapping peptides.
What information should be prepared before requesting a feasibility review?
The most useful inputs are sample type, buffer or formulation details, available amount, storage history, known purity concerns, prior peptide maps or vendor reports, and the minimum sequence confidence needed for your downstream use.
How to order?

