





Mass Spectrometry in Subcellular Proteomics: Organelle Fractionation, Localization, and Quantitative Analysis
- Subcellular proteomics depends on both fraction purity and MS depth, so sample preparation quality directly shapes biological interpretation.
- LC-MS/MS identifies organelle proteins, measures abundance changes, and can also support PTM and relocation analysis.
- Differential centrifugation, density gradients, immunoenrichment, and proximity labeling are common upstream enrichment strategies.
- Label-free, TMT/iTRAQ, SILAC, and targeted MS each fit different subcellular study designs.
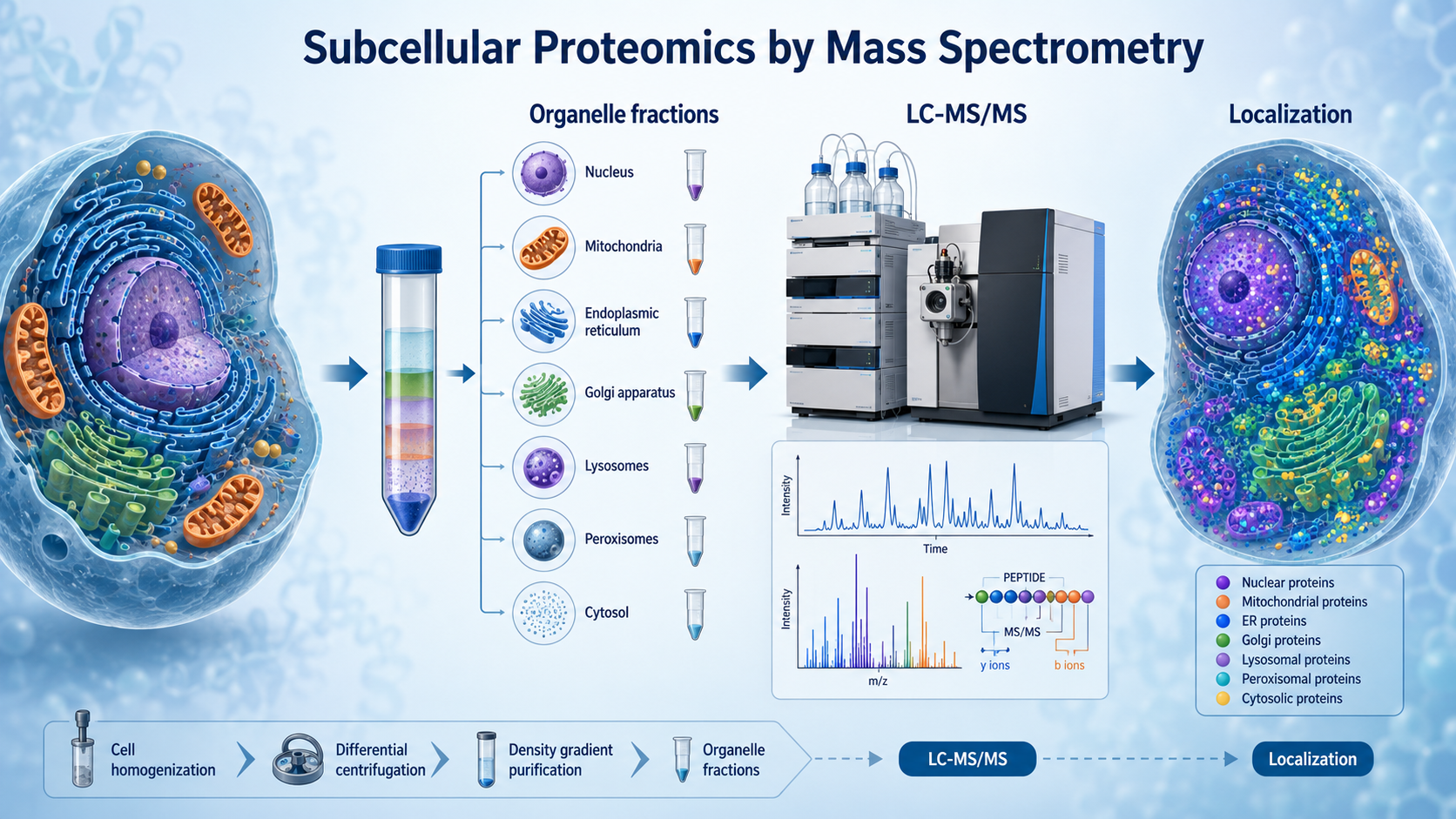
Mass spectrometry enables subcellular proteomics by identifying and quantifying proteins from purified organelles or compartment-enriched fractions after fractionation, peptide preparation, and LC-MS/MS analysis. Instead of asking only which proteins are present in a sample, subcellular proteomics asks where proteins reside, how they redistribute across compartments, and how organelle-specific proteomes change under disease, stress, treatment, or developmental transitions.
Key Takeaways
Why Subcellular Proteomics Matters?
Cells are organized into compartments such as mitochondria, lysosomes, Golgi, endoplasmic reticulum, nucleus, and plasma membrane domains. These compartments differ in protein composition, signaling logic, metabolic activity, and stress response.
Related Services
Subcellular Proteomics Service
Protein Subcellular Localization Service
Subcellular Structure and Organelle Proteomics Service
Subcellular Structures Protein Identification Service
Fractionation Comes First
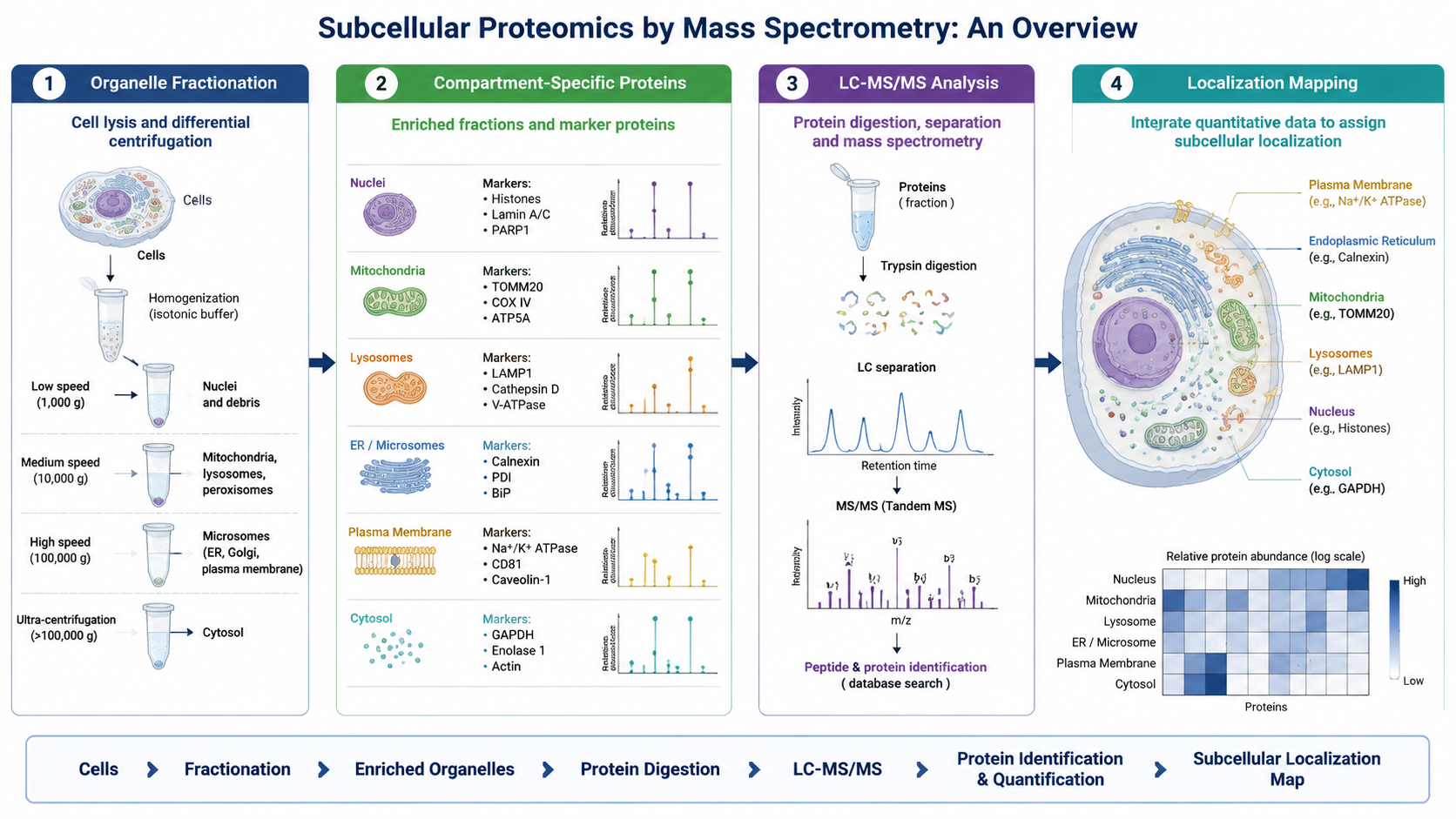
Mass spectrometry cannot resolve organelle localization if the fractionation step fails. Differential centrifugation and density gradients are widely used for mitochondria, ER, lysosomes, Golgi, and related fractions. Immunomagnetic enrichment helps when a membrane marker or organelle surface antigen is available.
How LC-MS/MS Analyzes Subcellular Fractions?
Once proteins are extracted from the fraction of interest, they are digested into peptides, separated by liquid chromatography, and analyzed by tandem mass spectrometry. MS1 scans detect precursor ions, while MS2 spectra support peptide identification and site-level modification analysis.
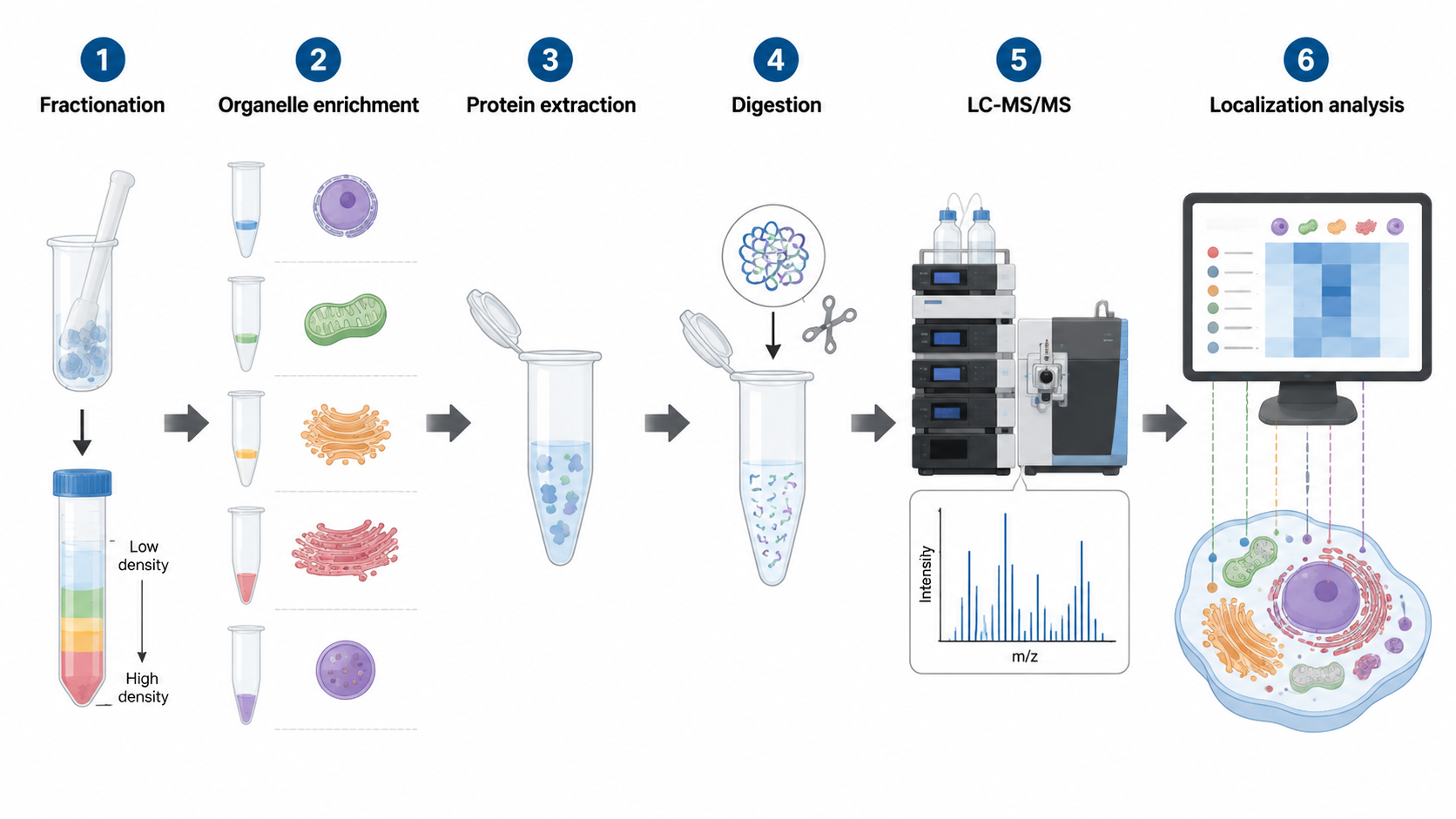
Quantitative Strategies in Subcellular Proteomics
1. Label-Free Quantification
Label-free workflows are flexible and fit large cohorts or cases where many fractionated samples must be processed without labeling chemistry.
2. TMT or iTRAQ
Multiplex labeling is useful when several organelle fractions, treatment groups, or time points need consistent relative comparison in a pooled experiment.
3. SILAC
SILAC is attractive in cell culture systems where stable metabolic labeling can support clean relative quantification across compartments.
Localization Precision and Contamination Control
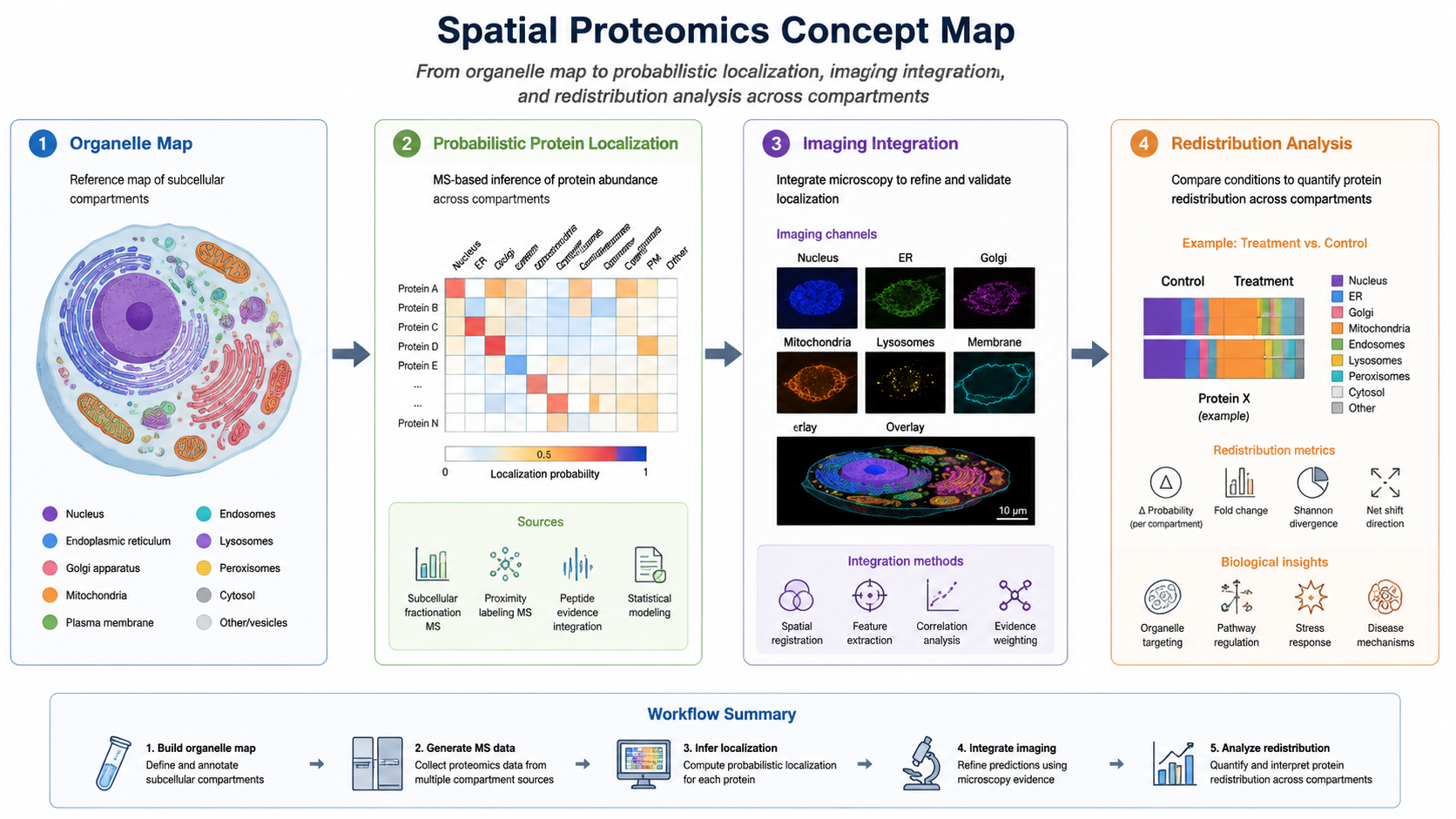
One of the biggest analytical challenges is distinguishing true compartment residents from contaminants or proteins that shuttle between compartments. Marker proteins, replicate concordance, enrichment scores, and probabilistic localization modeling all help.
Common Applications
| Use case | What subcellular MS reveals | Why it matters |
|---|---|---|
| Tumor heterogeneity | Organelle-specific metabolic rewiring | Explains adaptive stress states |
| Neurodegeneration | Golgi, lysosome, or mitochondrial remodeling | Connects localization to pathology |
| Drug mechanism studies | Protein redistribution across compartments | Reveals pathway-level effects |
| Signaling biology | Movement of pathway proteins between sites | Adds spatial context to activation logic |
Future Directions
Subcellular proteomics is moving toward higher spatial resolution, lower sample input, and stronger integration with imaging and multi-omics. Single-cell mass spectrometry, spatial omics, proximity labeling, and AI-assisted localization analysis are all expanding what can be learned about compartment-specific biology.
FAQ
1. How does mass spectrometry work in subcellular proteomics?
It analyzes peptides derived from purified or enriched cellular compartments, then identifies and quantifies proteins so researchers can infer organelle composition and protein redistribution.
2. Why is fraction purity so important?
Because contamination between organelles can create false localization claims, especially for abundant proteins that appear in multiple fractions through carryover.
3. Can subcellular proteomics detect protein relocation?
Yes. Comparing compartment-resolved abundance patterns across conditions can reveal redistribution events, especially when combined with marker controls and replicate statistics.
Conclusion
Mass spectrometry is the analytical core of subcellular proteomics, but the biology it reveals depends on strong fractionation design and careful contamination control. When organelle enrichment, LC-MS/MS analysis, quantitative strategy, and localization modeling are aligned, subcellular proteomics can reveal spatial protein behavior that whole-cell proteomics often misses.
How to order?

