





Protein Expression Profiling: Quantitative Proteomics Workflow, Applications, and Method Selection
-
Protein expression profiling measures protein-level changes that transcriptomics may not fully capture.
-
LC-MS/MS workflows can use label-free, DIA, TMT, iTRAQ, or targeted proteomics depending on throughput, precision, and study design.
-
Good profiling studies require consistent sample preparation, replicate planning, normalization, missing-value review, and batch effect control.
-
Differential protein expression can support biomarker discovery, drug target validation, toxicology, food safety, and mechanism-of-action studies.
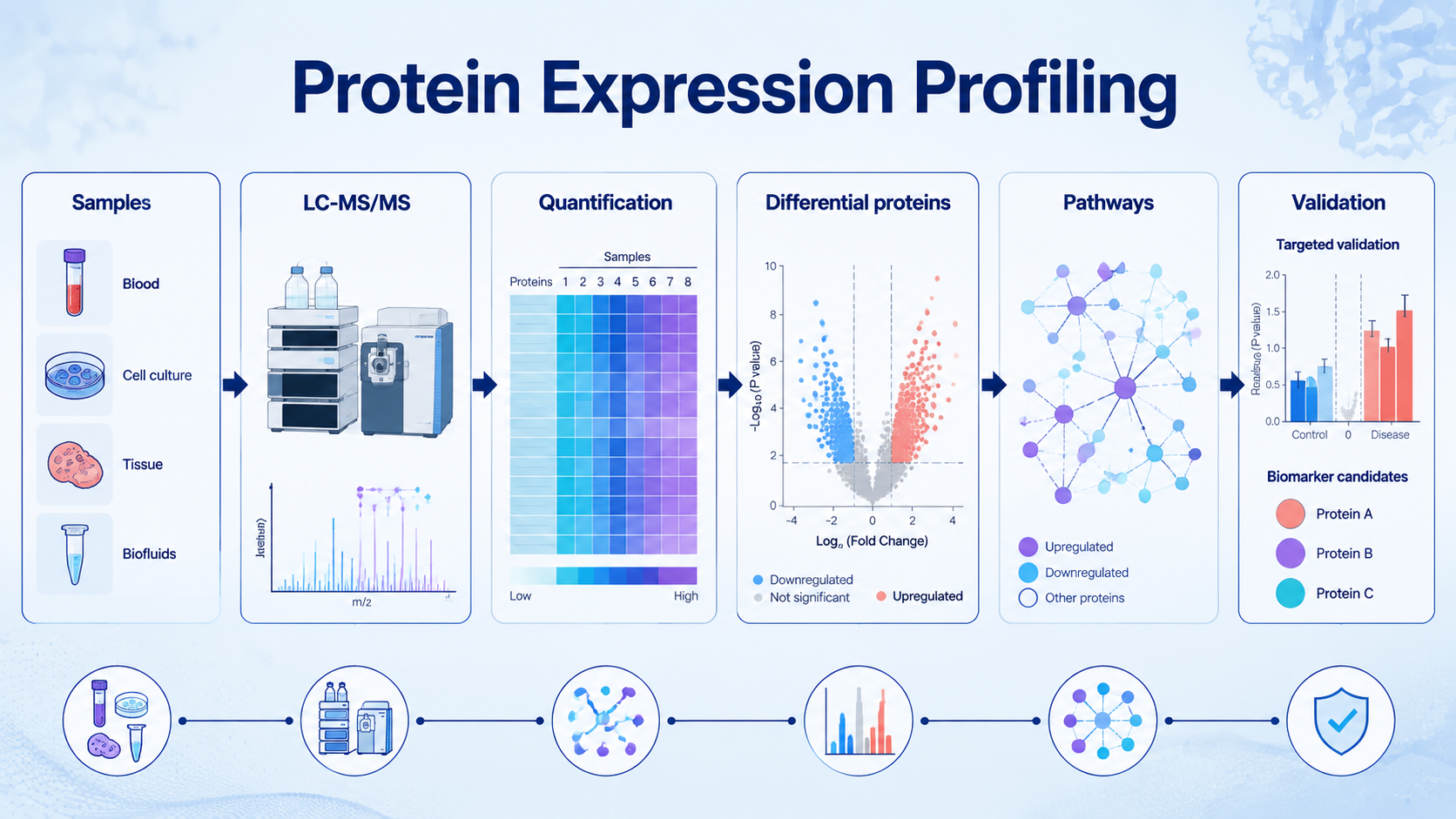
Protein expression profiling measures how protein abundance changes across cells, tissues, organisms, disease states, treatments, or environmental conditions. In proteomics, it is usually performed with LC-MS/MS to identify and quantify hundreds to thousands of proteins in parallel, then connect differential expression patterns to pathways, biomarkers, drug responses, or biological mechanisms.
Key Takeaways
What Does Protein Expression Profiling Measure?
Protein expression profiling measures relative or absolute protein abundance across sample groups. It can compare healthy and diseased tissues, treated and untreated cells, time-course samples, environmental exposure models, or production batches. The output is usually a quantified protein matrix with statistical comparisons, pathway annotations, and candidate proteins for follow-up validation.
Related Services
4D-DIA Quantitative Proteomics Service
DIA Quantitative Proteomics Analysis Service
4D Label-Free Quantitative Proteomics Service
Label Free Quantification Proteomics Service
Core Workflow
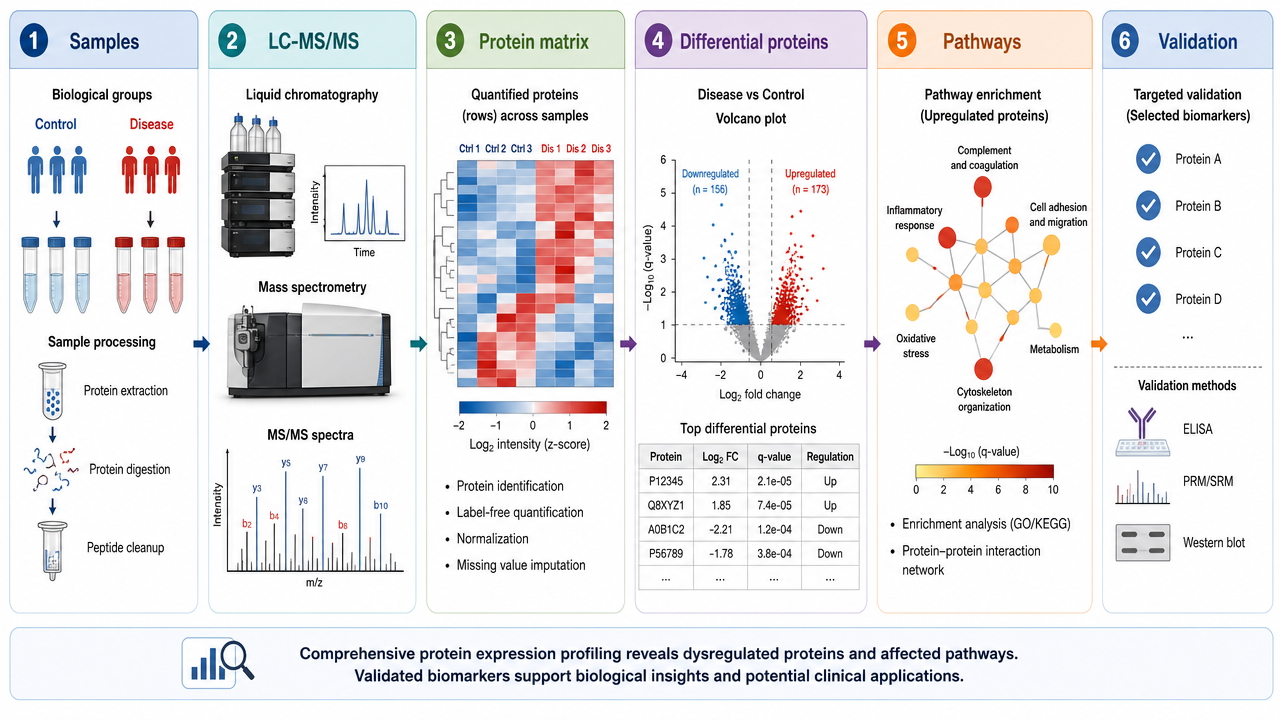
A typical protein expression profiling workflow includes sample collection, protein extraction, digestion, peptide cleanup, LC-MS/MS acquisition, database search or DIA analysis, protein quantification, normalization, statistical testing, pathway analysis, and candidate validation.
For complex samples, the most fragile parts are usually upstream: inconsistent extraction, poor digestion efficiency, sample loss, and batch effects can become visible later as false differential expression.
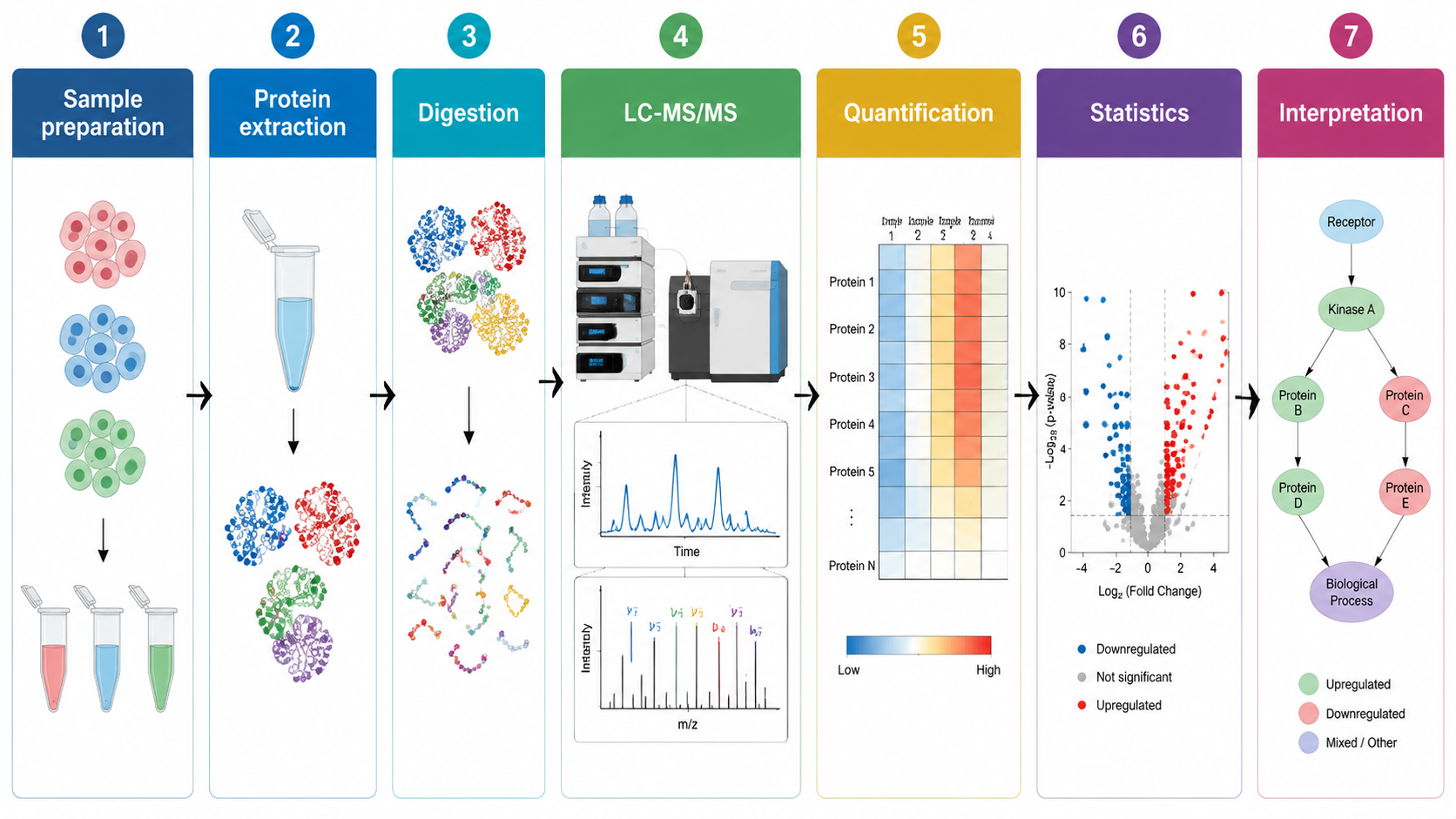
Common Acquisition Strategies
Label-free proteomics is useful when many samples need broad profiling without chemical labeling. DIA improves quantitative consistency across larger cohorts by systematically fragmenting peptide windows. TMT and iTRAQ support multiplexed comparisons, but require careful control for ratio compression and batch structure. PRM or SRM can then validate selected proteins with targeted sensitivity.
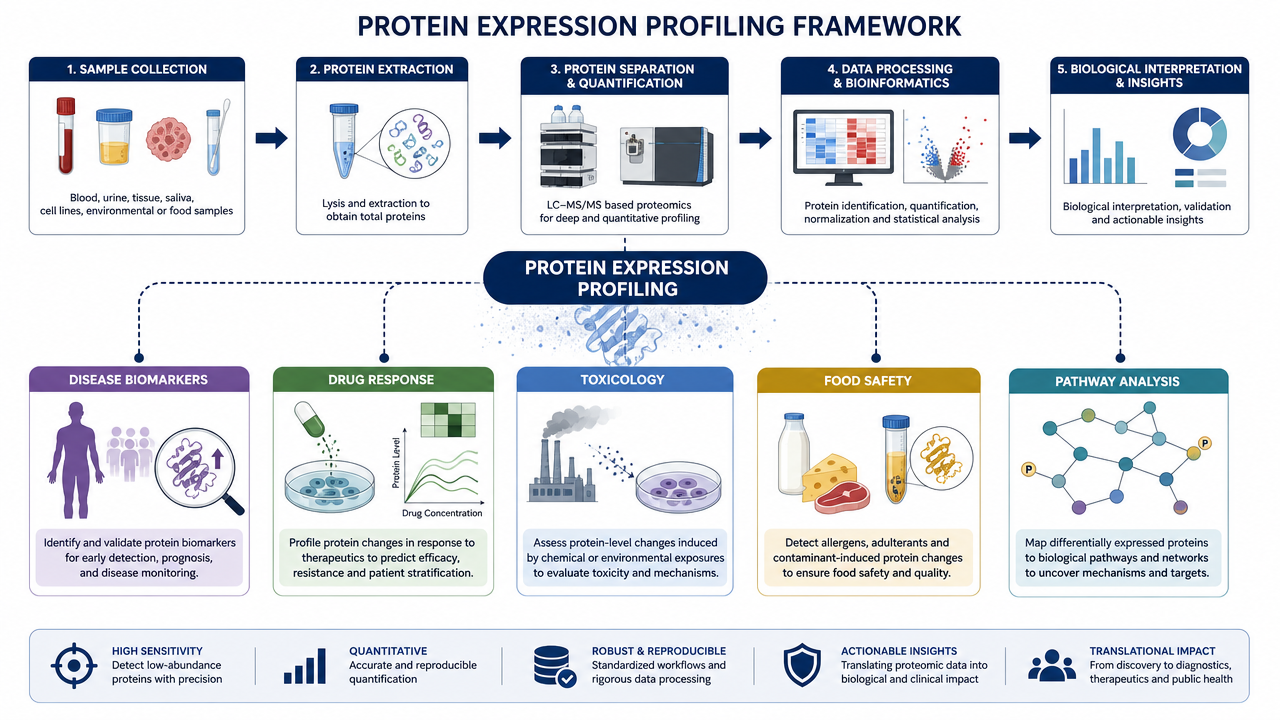
Applications
1. Disease Biomarker Discovery
By comparing diseased and healthy samples, protein expression profiling can reveal candidate biomarkers linked to diagnosis, prognosis, disease subtype, or treatment response.
2. Drug Target and Mechanism Studies
Profiling treated and untreated cells or tissues can show whether a compound affects the intended pathway, triggers compensatory signaling, or creates off-target effects.
3. Toxicology and Environmental Response
Exposure models can be profiled to identify stress response proteins, detoxification pathways, inflammation markers, or organism defense mechanisms.
4. Food Safety and Quality Research
Protein expression profiling can help identify allergens, bacterial toxins, processing-related protein changes, or quality markers in food samples.
Method Selection
| Study Goal | Recommended Method | Main Strength | Main Caution |
|---|---|---|---|
| Broad discovery with many samples | Label-free quantitative proteomics | Flexible and cost-effective | Missing values require review |
| Cohort-scale consistency | DIA proteomics | Reproducible quantification | Requires optimized analysis workflow |
| Multiplexed comparison | TMT or iTRAQ | Many samples in one batch | Ratio compression can reduce effect sizes |
| Low-abundance validation | PRM or SRM | Targeted sensitivity | Limited to selected proteins |
FAQ
1. What is protein expression profiling?
Protein expression profiling is the measurement of protein abundance patterns across biological samples, conditions, or time points to identify proteins and pathways that change.
2. Why use proteomics instead of only transcriptomics?
Protein abundance is affected by translation, degradation, secretion, localization, and post-translational regulation. These layers are not fully captured by RNA measurements.
3. Which method is best for protein expression profiling?
The best method depends on sample number, expected effect size, budget, and depth needs. Label-free and DIA workflows are common for discovery studies, while PRM or SRM is often used for targeted validation.
4. How are differentially expressed proteins validated?
Candidates can be validated by targeted MS, immunoblotting, ELISA, activity assays, independent cohorts, or orthogonal omics evidence.
Conclusion
Protein expression profiling is most useful when the experimental design is as strong as the mass spectrometry. Reliable results depend on clean sample handling, the right quantitative method, transparent statistics, and validation of the proteins that drive the biological story.
How to order?

