





Overview of Protein Post-Translational Modifications (PTMs)
-
SUMO (Small Ubiquitin-related Modifier) – fine-tunes transcription-factor activity, DNA repair, and nucleocytoplasmic transport.
-
NEDD8 – modifies Cullin proteins to regulate the activity of Cullin–RING E3 ligase (CRL) complexes.
-
ISG15 – up-regulated during antiviral responses to amplify interferon signaling.
-
Enrichment with high-affinity K-ε-GG antibodies that specifically capture di-glycine–containing peptides.
-
Acquisition of high-resolution MS/MS spectra to precisely localize the modified residue via b/y-ion fragments.
-
Multi-enzyme digestion strategies (e.g., trypsin plus Lys-C) to boost sequence coverage and uncover additional modified sites.
While proteomics has provided us with comprehensive maps of protein expression within cells, information solely about protein expression levels is insufficient to explain the dynamic regulatory behaviors of biological systems. The functional state of a protein is not simply determined by its expression level but is highly dependent on the covalent modifications that occur post-translationally, known as post-translational modifications (PTMs). These modifications involve the enzymatic or non-enzymatic addition of chemical groups (such as phosphate, acetyl, methyl, ubiquitin, etc.) to specific amino acid residues, thereby regulating the protein's conformation, enzymatic activity, stability, subcellular localization, and interactions with other macromolecules.
Current studies show that more than 80% of functional proteins in the body are in some form of post-translationally modified state. Many of these modifications are dynamic, reversible, and spatially and temporally specific, allowing them to respond to external stimuli within seconds to minutes. For instance, in cellular signal transduction, the coordinated actions of kinases and phosphatases enable amplification and feedback regulation of signaling cascades. Meanwhile, the ubiquitin-proteasome system, through variations in polyubiquitin chain types, creates complex selective degradation and non-degradation signaling pathways. More importantly, multiple modifications often do not occur independently but are interconnected in a process known as "crosstalk." This crosstalk involves interactions, competition, and cooperation between different modifications, forming a highly complex "modification code (PTM code)" on the protein surface that profoundly impacts cellular fate decisions and functional differentiation.
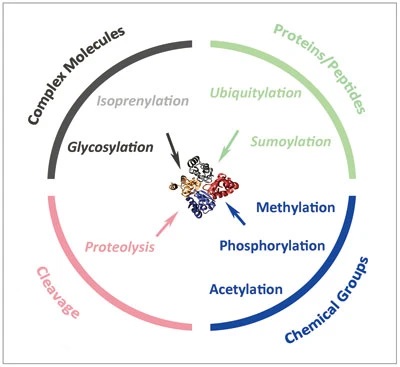
Cell Res 24, 143–160 (2014).
Multiple reversible and irreversible protein post-translational modification (PTM) mechanisms in eukaryotic cells
From a systems biology perspective, PTMs are considered a key bridge linking genotype to phenotype, representing a fourth layer of regulatory logic beyond expression levels. Their study not only unveils the intricate mechanisms underlying cellular signaling networks, epigenetic regulation, and stress responses but also provides crucial breakthroughs for understanding disease mechanisms, drug target identification, and the discovery of clinical biomarkers. In recent years, the discovery of novel modifications (such as lactylation, butyrylation, and glutathionylation) has further expanded our understanding of the connectivity between metabolism, epigenetics, and signaling. This highlights that PTMs are one of the core entry points for understanding the complexity of biological systems.
Common Types of Post-Translational Modifications (PTMs)
Protein post-translational modifications (PTMs) encompass hundreds of different types of chemical modifications, some of which are highly conserved in evolution, while others are restricted to specific physiological states or particular species. These modifications primarily occur on the side-chain functional groups of the protein backbone (especially on residues such as Ser, Thr, Tyr, Lys, Arg, Cys, etc.), where small molecular groups, peptides, or glycan chains are introduced through enzymatic or non-enzymatic processes. Below are some of the most representative PTM types, their mechanisms, and their roles in research and clinical applications:
1. Phosphorylation
Phosphorylation is the most extensively studied PTM. Protein kinases catalyze the transfer of a phosphate group from ATP to serine (Ser), threonine (Thr), or tyrosine (Tyr) residues on target proteins, generating phosphopeptides. This modification is highly dynamic and reversible: protein phosphatases can promptly remove the phosphate group, creating an on/off regulatory system.
Phosphorylation underlies the vast majority of intracellular signaling cascades. In canonical pathways such as MAPK, PI3K–Akt, and JAK–STAT, discrete phosphorylation events drive kinase activation, promote nuclear translocation of transcription factors, and govern cell-cycle progression. Because of its rapid kinetics and strong signal-amplification capacity, phosphorylation is widely regarded as the central information-transduction modification in cellular biology.
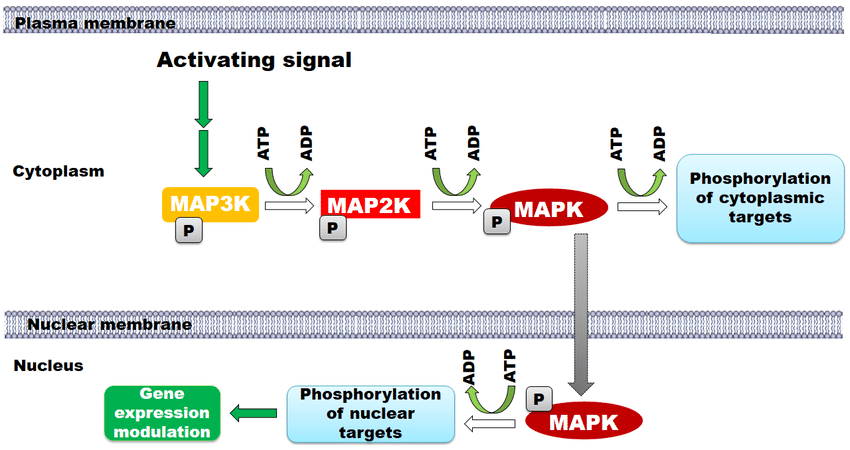
PLoS One. 2016 May 31;11(5).
A schematic representation of the MAPK cascade and its activation mechanisms
In mass-spectrometry workflows, phosphopeptides carry a net negative charge and therefore must be selectively enriched—typically with immobilized-metal affinity chromatography (IMAC) or TiO₂—before analysis. Site localization then relies on high-resolution MS/MS data, where characteristic neutral-loss fragments (e.g., –98 Da) and precise matching of b/y ions together pinpoint the modified residue.
2. Acetylation
Acetylation was first discovered on histones but is now known to be widespread on non-histone proteins as well. By neutralizing the positive charge of lysine side chains, the modification alters electrostatic interactions between proteins and DNA, or between protein partners. In the histone context, this charge modulation directly dictates chromatin openness and, consequently, transcriptional accessibility—making lysine acetylation a foundational mark of epigenetic regulation.
More recently, non-histone acetylation has become a focal point of research. In metabolic enzymes, signaling mediators, and structural proteins, acetylation influences conformational stability, catalytic activity, and even subcellular localization. A classic example is the tumor suppressor p53: multi-site acetylation not only enhances its DNA-binding affinity but also modulates crosstalk with the ubiquitination machinery, prolonging p53 half-life and preventing aberrant degradation.
The modification is tightly coupled to cellular energy metabolism. Fluctuations in intracellular acetyl-CoA levels can directly shift global acetylation status, creating a conduit that links metabolic flux to epigenetic landscapes. Acetylation, therefore, is no longer viewed merely as a “transcriptional switch” but rather as a metabolically driven epigenetic mediator.
Mass-spectrometry considerations: Because lysine acetylation does not change peptide charge state, acetylated peptides are harder to recognize in complex mixtures. High-affinity anti-acetyl-Lys antibodies are commonly used for enrichment, and digestion with Lys-N or trypsin helps preserve critical modification sites. Quantification is typically performed with TMT labeling or label-free approaches. During data analysis, special care is required to avoid misassignments caused by mass shifts that are close to other modifications such as formylation or glutarylation.
3. Methylation
Unlike phosphorylation or acetylation, methylation does not alter the net charge of a protein; instead, it generally establishes a “stable state” of regulation. Histone methylation is the most intensively characterized example. Both the residue identity and the methylation stoichiometry—mono-, di-, or tri-methylation—precisely shape chromatin structure and gene expression. For example, H3K4me3 is widely recognized as a promoter-activation mark, whereas H3K27me3 is strongly associated with transcriptionally silent regions.
The influence of methylation extends far beyond histones. Methylation of transcription factors, signaling proteins, and RNA-binding proteins is increasingly implicated in cell development, embryonic differentiation, and stem-cell fate decisions. Notably, the biological roles of many methyl-lysine or methyl-arginine sites remain undefined; some methyl marks may serve as “protective caps” rather than activators, underscoring that our functional interpretation of protein methylation is still at an early stage.
Mass-spectrometry considerations: Methylation adds only +14.0157 Da per methyl group, so distinguishing mono-, di-, and tri-methyl states demands high mass accuracy. Spectral overlap among isomeric methylated peptides is substantial; therefore, high-resolution MS, complementary protease digests, and enrichment with anti-methyl-lysine/arginine antibodies are recommended to achieve confident site identification and quantification.
4. Ubiquitination and Ubiquitin-like Modifications
Ubiquitination is the covalent attachment of the 76-amino-acid protein ubiquitin to a lysine side chain on the target protein through an isopeptide bond. Among all PTMs, ubiquitination is one of the most “versatile.” In addition to acting as a direct tag for proteasomal degradation, it regulates numerous cellular processes by assembling poly-ubiquitin chains of distinct linkages (K48, K63, K11, linear, and others) that modulate cell signaling, DNA repair, membrane trafficking, and even transcriptional control.
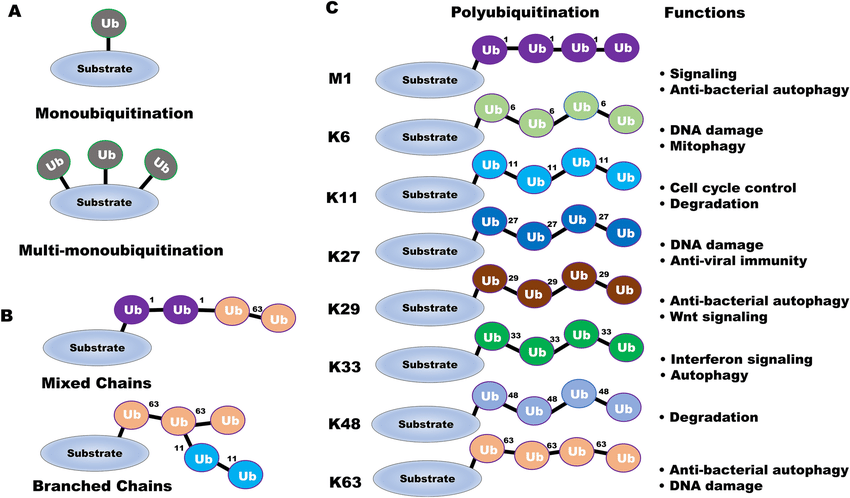
Apoptosis. 2022 Oct; 27(9-10):668-684.
Various types of ubiquitin linkage and related functions
Beyond ubiquitin itself, cells express several ubiquitin-like proteins (Ubls) that share a similar β-grasp fold but serve different roles:
Because ubiquitin is an intact protein rather than a small chemical group, tryptic digestion leaves a characteristic di-glycine (Gly-Gly) remnant on the modified lysine, producing a +114.0429 Da mass shift. Confident identification of ubiquitination sites therefore hinges on:
5. Glycosylation
Glycosylation is the only PTM whose biological activity is fundamentally rooted in structural diversity. In contrast to “single-point” functional marks such as phosphorylation or acetylation, the impact of glycosylation depends on the composition, length, branching pattern, and site-specific presentation of its carbohydrate chains.
N-linked (Asn) and O-linked (Ser/Thr) glycosylation are ubiquitous on cell-surface, secreted, and receptor proteins, where they influence folding efficiency, membrane localization, and immune evasion. A prominent example is PD-L1: N-glycosylation enhances the ligand’s stability and surface expression, contributing to tumor-mediated immune suppression. Changes in glycan structures are also hallmarks of oncogenic transformation; for instance, the Tn antigen (O-GalNAc) is up-regulated in many cancer cells.
The principal analytical challenge is heterogeneity: a single glycosylation site can carry dozens of distinct glycoforms, imposing immense demands on detection and quantification. Current mainstream workflows combine hydrophilic interaction liquid chromatography (HILIC) or porous graphitized carbon (PGC) separations with MSⁿ techniques, yet fully resolving complex glycan architectures often requires multi-platform data integration.
Glycosylation studies are therefore highly dependent on bioinformatic annotation. Tools such as GlycoWorkbench and Byonic, which embed curated glycan-structure databases, are indispensable—particularly in untargeted analyses—to ensure confident identification and accurate glycoform assignment.
6. Emerging Modifications
Advances in high-resolution mass spectrometry (HR-MS) and targeted-enrichment technologies have brought many previously elusive PTMs into view. These modifications are often derived directly from metabolic intermediates and are characterized by rapid kinetics, broad regulatory scope, and high sensitivity to environmental change. They now appear to be pivotal nodes that connect cellular metabolic states with epigenetic programs. Below is a representative selection of novel modifications currently attracting intense research interest.
(1) Lactylation (Kla)
Lysine lactylation (Kla) is the archetype of metabolite-driven PTMs. First reported by Zhang et al. in 2019 on histone H3, the modification involves transfer of an acyl group from endogenous L-lactate to the ε-amino group of lysine. Kla levels rise sharply under hypoxia, heightened glycolysis (Warburg effect), and immune activation, and have been shown to influence macrophage polarization (M1 → M2), stem-cell fate decisions, and oncogene expression.
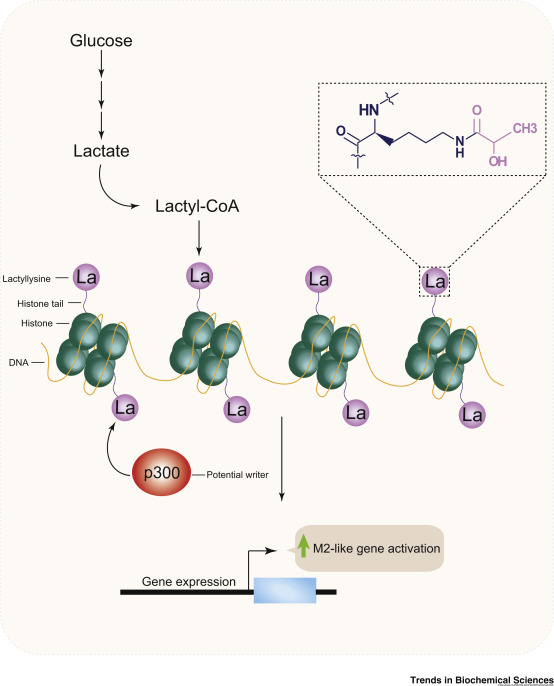
Trends Biochem Sci. 2020 Mar;45(3):179-182.
Histone lactylation mediates gene expression to promote an M2-like phenotype
Mechanistically, lactate emerges not merely as a metabolic by-product but as a direct signaling molecule capable of modulating chromatin activity. Non-histone targets—including metabolic enzymes and transcription factors—have also been identified, indicating that lactylation exerts broad control over transcriptional regulation and protein-interaction networks.
Analytical challenges: Kla introduces a +72.0211 Da mass shift that can be confused with other acyl modifications, and modified peptides are typically of very low abundance. Reliable detection therefore requires enrichment with highly specific anti-Kla antibodies coupled to nano-LC–MS/MS analysis at high resolution.
(2) β-Hydroxybutyrylation (Kbhb)
β-Hydroxybutyrate (BHB) is the predominant ketone body generated during starvation, prolonged fasting, or ketogenic feeding. When cellular BHB concentrations rise, the metabolite serves as an acyl donor that modifies lysine ε-amino groups, creating β-hydroxybutyrylation marks. Kbhb is abundant in liver, skeletal muscle, and brain, and is viewed as a hallmark of epigenetic transcriptional reprogramming under energy-deficit conditions.
Functional studies show that Kbhb can compete with lysine acetylation, thereby suppressing the expression of inflammatory genes (e.g., NF-κB targets) and enhancing resistance to oxidative stress—traits that place the modification at the center of research on longevity and metabolic health.
Analytical challenges:Multiple structural isomers of Kbhb exist. High-resolution MS, complemented by detailed tandem-MS fragmentation patterns, is required for unambiguous site assignment. For accurate quantification, stable-isotope labeling (SILAC) or targeted methods such as PRM are strongly recommended.
(3) S-Glutathionylation
Glutathione (GSH) is the predominant intracellular antioxidant. Under oxidative challenge, it forms mixed disulfides with protein thiols, a reversible modification known as S-glutathionylation. By shielding critical cysteine residues from irreversible oxidation, S-glutathionylation preserves enzyme function, modulates catalytic activity, and propagates reactive-oxygen–species (ROS) signaling. The modification targets cysteine side chains and can be removed by glutathione reductase (GR) or the thioredoxin (Trx) system.
Mass-spectrometric considerations: The glycine–cysteine–glutamate adduct introduces a +305.0682 Da shift. Because thiols are highly reactive, samples must be processed rapidly and at low temperature to avoid artifactual modifications. Derivatization-based capture strategies—such as biotin-labeled GSH probes—are increasingly employed to enrich low-abundance S-glutathionylated peptides for downstream LC–MS/MS analysis.
(4) Acetylglutarylation, Propionylation, Butyrylation, and Related Acyl Marks: Signatures of Mitochondrial Lipid Metabolism
Acyl modifications derived from fatty-acid intermediates—including acetylglutarylation, propionylation, and butyrylation—are prevalent on mitochondrial inner-membrane proteins and metabolic enzymes. Their occurrence mirrors cellular lipid-metabolic activity and, by competing with lysine acetylation, influences transcription-factor function, chromatin architecture, and cell-fate decisions. For example, lysine butyrylation has been reported to modulate p53-dependent apoptotic pathways.
Analytically, these marks pose twin challenges: extensive site overlap and mass shifts that closely resemble one another. Confident identification therefore demands a combination of modification-specific enrichment chemistries and targeted fragmentation (e.g., neutral-loss monitoring or diagnostic ions) in high-resolution MS workflows.
How to order?

