





Mass Spectrometry-Based Quantitative Proteomics: Labeled and Label-Free Quantification Strategies
The core objective of quantitative proteomics is to accurately measure protein abundance across different experimental conditions using mass spectrometry (MS). Thanks to its high resolution and sensitivity, MS allows for the precise quantification of protein expression levels and enables the simultaneous analysis of multiple samples. To achieve this, MS is typically integrated with various labeling strategies. Among the most commonly used strategies in MS-based proteomics are label-free quantification and labeled quantification.
1. Label-Free Protein Quantitative Proteomics
Label-free quantification refers to quantification methods that do not rely on any chemical or isotopic labeling. Protein abundance is instead calculated based on peptide signal intensity or spectral counting. The most widely used LFQ approaches include intensity-based quantification and spectral counting.
1.1 Signal Intensity-Based Quantification
Signal intensity-based quantification measures the intensity of peptide ions detected in the mass spectrometer, under the assumption that peptide ion intensity is proportional to its abundance. In this approach, proteins are enzymatically digested into peptides, separated via liquid chromatography, and analyzed by MS. The signal intensity of each peptide reflects its relative abundance, which is then used to estimate changes in protein expression between samples. This method is straightforward, cost-effective, and well-suited for large-scale and complex sample analysis. However, its accuracy can be affected by differences in ionization efficiency and sample complexity.
1.2 Spectral Counting
Spectral counting estimates protein abundance by counting the number of MS/MS spectra matched to peptides from each protein. During MS analysis, proteins are digested into peptides, which are then fragmented and detected as spectra. The number of spectra identified for a given protein correlates with its abundance. Spectral counting offers simplicity and high throughput and is commonly used in relative quantification. However, it has several limitations: it is sensitive to ionization efficiency, less accurate for low-abundance proteins, and cannot provide absolute quantification. Additionally, its accuracy is highly dependent on the completeness of the reference database.
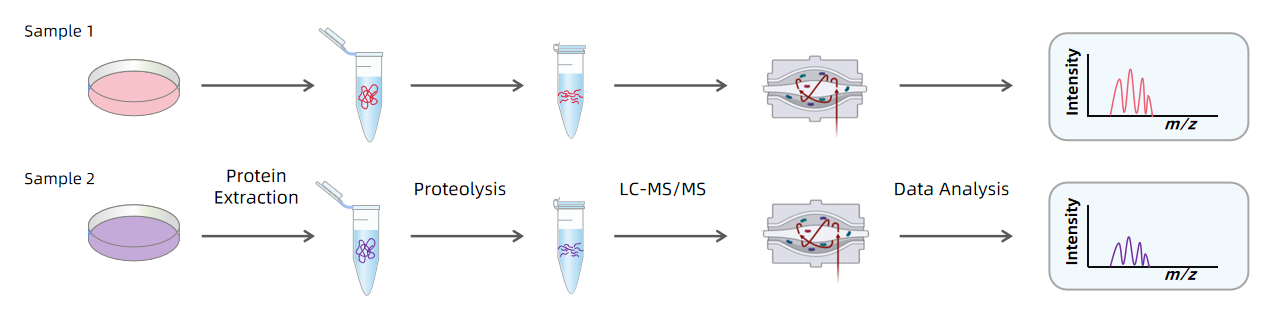
Figure 1. Workflow of Label-free Quantitative Proteomics
1.3 4D Label-free Quantitative Proteomics
4D Label-Free Quantitative Proteomics integrates ion mobility spectrometry (IM) and data-independent acquisition (DIA) for high-resolution, high-throughput protein quantification. This method incorporates four-dimensional separation—retention time, mass-to-charge ratio (m/z), ion intensity, and ion mobility—to improve selectivity and resolution. It is typically performed on instruments like the Bruker timsTOF Pro using the diaPASEF (Parallel Accumulation–Serial Fragmentation) acquisition method. The workflow includes protein extraction, digestion, LC separation, ion mobility separation, MS detection, and downstream bioinformatics analysis. It does not require any labeling, making it compatible with a wide range of biological sample types.
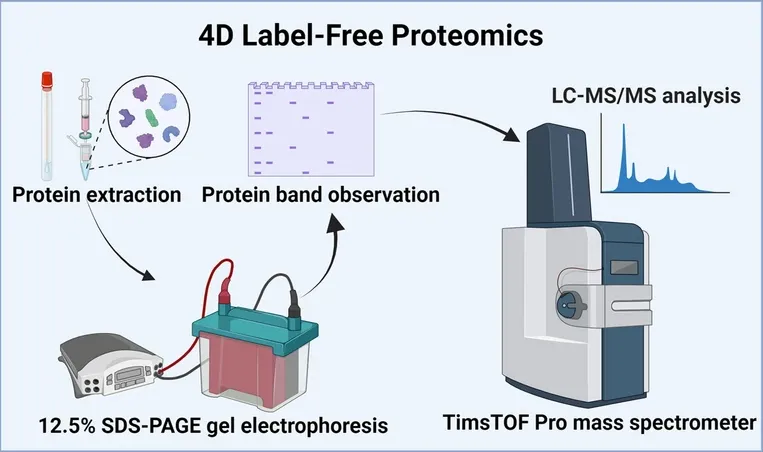
Jiang, H. et al. BMC Pregnancy and Childbirth, 2024.
Figure 2. Workflow of 4D Label-free Quantitative Proteomics
This method significantly enhances the identification of low-abundance proteins and structurally similar peptides, improves quantification accuracy, and increases reproducibility. Its label-free nature ensures lower cost and simpler workflows, making it suitable for large-scale studies. However, 4D-LFQ requires specific instrumentation and robust data analysis capabilities, and it may be less effective for batch-to-batch comparisons than TMT or iTRAQ. It is ideal for in-depth basic research and dynamic proteome profiling, rather than standardized clinical applications.
2. Label-Based Protein Quantitative Proteomics
After discussing the advantages and challenges of label-free quantification, we now turn to labeled quantification strategies, which rely on chemical or isotopic tags to quantify protein abundance. These strategies typically offer higher accuracy and are well-suited for comparative analysis across multiple samples.
2.1 SILAC (Stable Isotope Labeling by Amino Acids in Cell Culture)
SILAC is a metabolic labeling technique that introduces stable isotope-labeled amino acids into cultured cells to label proteins during synthesis. By supplementing cell culture media with isotope-labeled lysine or arginine (e.g., ^15N-labeled), proteins naturally incorporate these labels. Labeled and unlabeled peptides can then be distinguished by their mass differences in MS. SILAC enables highly accurate quantification and is particularly useful for analyzing low-abundance proteins in complex biological matrices.
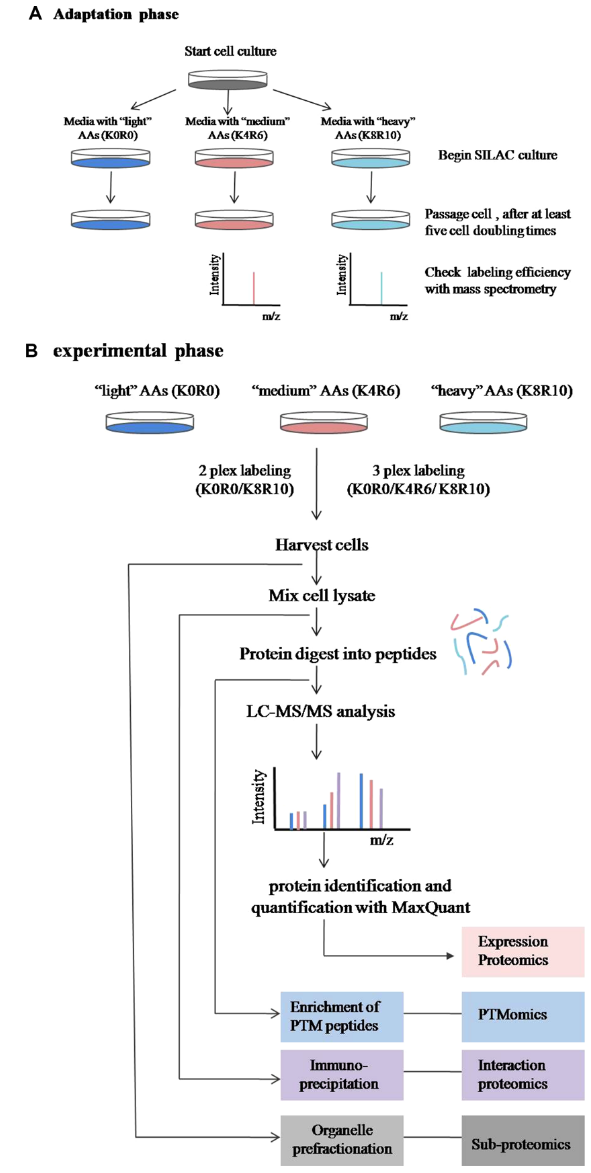
Chen, X. et al. Proteomics. 2015
Figure 3. Workflow for Quantitative Proteomic Experiments Using SILAC
SILAC offers high sensitivity and precision and is widely applicable to various biological models. However, it has limitations such as higher reagent cost and the need for high-performance MS platforms and data processing capabilities.
2.2 iTRAQ ( Isobaric Tags for Relative and Absolute Quantification)
iTRAQ is a chemical labeling technique that uses isobaric tags to enable the relative and absolute quantification of proteins across multiple samples. Each iTRAQ tag contains a reporter group and a reactive group. During MS/MS analysis, the tags fragment to release unique reporter ions, whose intensities reflect the abundance of peptides from different samples. iTRAQ allows simultaneous analysis of up to 8 samples in a single run, significantly improving throughput.Isobaric Tags for Relative and Absolute Quantification
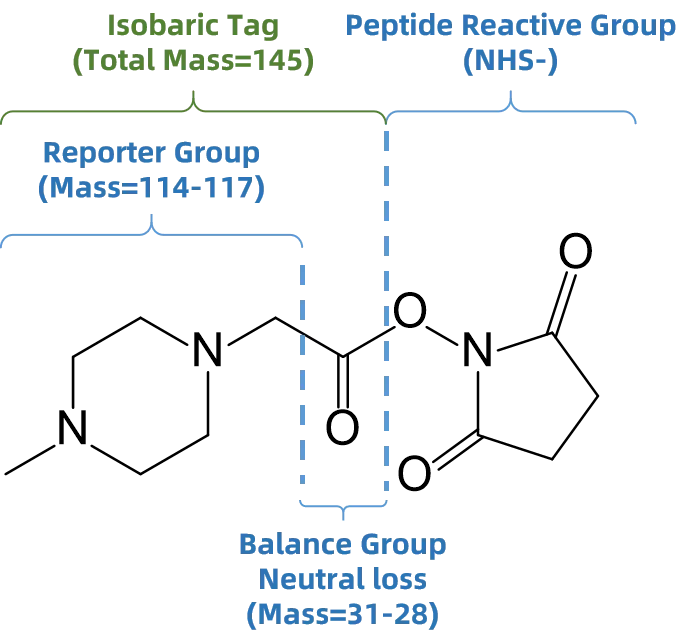
Figure 4. Structure of iTRAQ Tag
iTRAQ offers high accuracy and reproducibility due to consistent MS1 mass values across labeled peptides and the use of distinct MS2 reporter ions for quantification. It is widely used in disease research, drug development, and biomarker discovery. However, iTRAQ is relatively expensive and may suffer from reporter ion interference in complex samples, potentially affecting quantification precision. Proper labeling and sample preparation are essential for reliable results.
2.3 TMT (Tandem Mass Tags)
TMT is another isobaric labeling technology used for relative quantification. It labels peptides from different samples with tags of identical mass. During MS/MS fragmentation, these tags release distinct reporter ions, enabling quantification based on ion intensity. Like iTRAQ, TMT provides accurate and reproducible relative quantification across multiple samples.
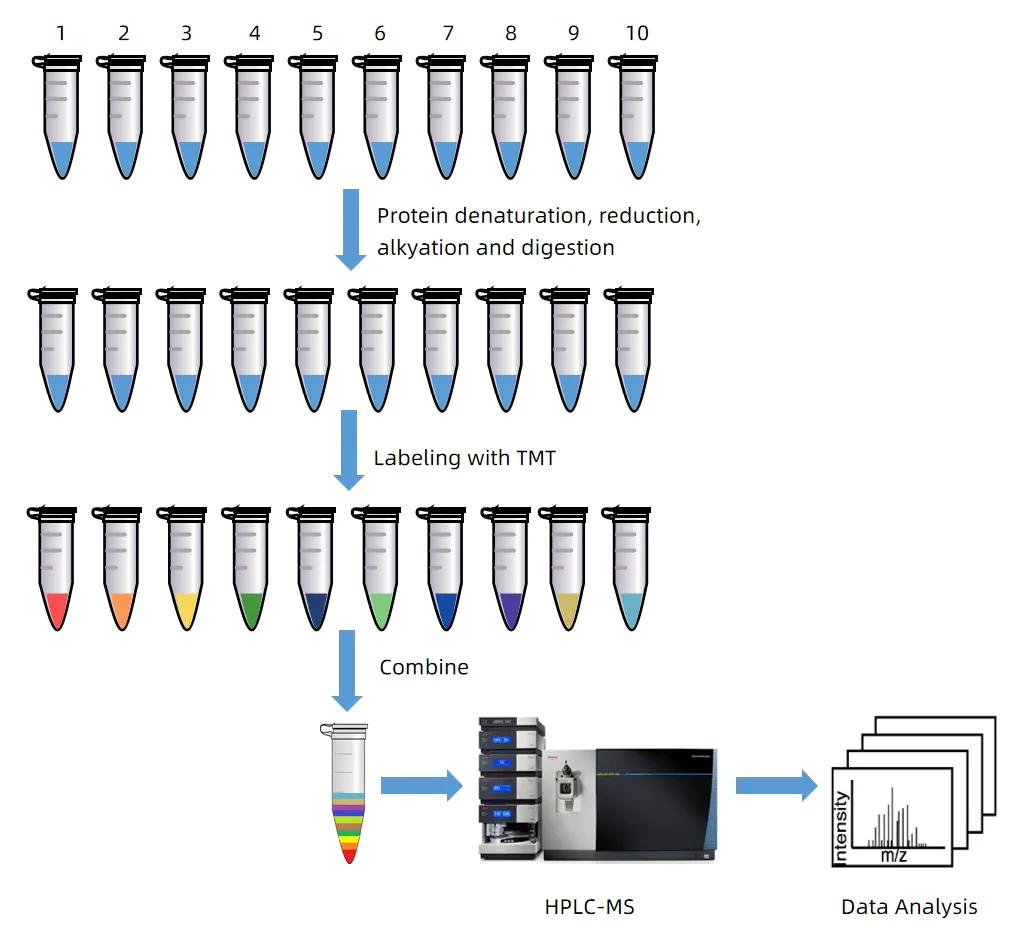
Figure 5. Workflow for Relative Quantification Using 10-plex TMT
TMT enables simultaneous analysis of up to 16–18 samples (TMTpro), making it highly suitable for large-scale comparative studies. It offers high precision and improved data consistency. However, the cost of TMT reagents and the complexity of sample processing are potential limitations. Interference between reporter ions may occur, especially in highly complex samples, which may affect quantification accuracy.
Quantitative proteomics has become an indispensable tool in modern life sciences, supporting breakthroughs in basic research, clinical diagnostics, and drug development. Both label-free and labeled quantification strategies offer unique advantages and challenges, and selecting the appropriate approach is critical for research accuracy and efficiency. As technologies continue to evolve, quantitative proteomics will play an increasingly vital role in precision medicine and biomarker discovery.
About Us
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider. We offer high-quality quantitative proteomics solutions, including label-free and labeled quantification strategies, tailored to meet the diverse needs of researchers worldwide. Our quantitative proteomics platform supports DIA, DDA, PRM, and MRM acquisition modes. If you have any questions or require expert guidance in proteomics, feel free to contact us—we are here to help.
Related Services
How to order?

