





Quantitative Proteomics Analysis Service
Quantitative proteomics precisely analyze protein expression differences and dynamic changes in samples using both label-based methods (such as SILAC, TMT, and iTRAQ) and label-free quantification strategies. Label-based proteomics leverage isotopic tags for simultaneous quantification across multiple samples, offering high sensitivity and reproducibility, while label-free methods require no complex preprocessing and are more suitable for large-scale sample analysis. These approaches serve as powerful tools for unraveling the molecular mechanisms of biological systems. Quantitative proteomics is essential for identifying potential biomarkers and understanding drug treatment mechanisms, revealing patterns of protein expression and modifications to facilitate early disease diagnosis, precision therapy, and innovative drug development.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider, provides advanced proteomics, metabolomics, and biopharmaceutical analysis services to researchers in biochemistry, biotechnology, and biopharmaceutical fields. Having extensive expertise, we offer a high-quality quantitative proteomics analysis service based on advanced mass spectrometry platforms to meet our clients' proteomics analysis needs.
Service at MtoZ Biolabs
Currently, quantitative proteomics techniques are primarily divided into label-based and label-free strategies. Label-based strategies include in vivo labeling (such as SILAC) and in vitro labeling (such as iTRAQ, TMT, and Dimethyl). Label-free strategies are further categorized into spectral counting and peptide ion intensity-based quantification.
1. Label-based Quantitative Proteomics
1. Quantitative Proteomics by SILAC
In vivo metabolic labeling methods incorporate stable isotopes into proteins through metabolic incorporation into living systems, such as stable isotope labeling by amino acids in cell culture (SILAC) and ¹⁵N labeling. For SILAC, cells are cultured in light or heavy culture media, which offering natural or stable isotope-labeled amino acids. After MS analysis, quantification is achieved by comparing the light/heavy peptide pairs at the MS1 level. SILAC minimizes technical variability by combining light-labeled and heavy-labeled samples early in the sample preparation workflow. However, in vivo metabolic labeling can only be applied to studies involving cell culture or model organisms, as samples must grow in customized media to incorporate stable isotopes during growth. Additionally, most in vivo metabolic labeling methods are limited to comparing 2-3 samples. Nevertheless, 5-plex SILAC experiments can be achieved using five different forms of arginine or a combination of two 3-plex SILAC experiments with shared experimental conditions.
2. Quantitative Proteomics by TMT
In vitro labeling methods, such as Tandem Mass Tags (TMT), chemically label proteins or peptides after sample extraction, enabling simultaneous quantification of multiple samples in a single experiment. TMT uses isotopically labeled reagents to tag peptides, generating reporter ions at the MS2 stage of mass spectrometry analysis. Quantification is achieved by comparing the intensities of these reporter ions, allowing for accurate relative quantification across samples. TMT significantly reduces technical variability by processing all samples together in a unified experimental workflow. Unlike in vivo labeling methods, TMT is not restricted to cell cultures or model organisms and can be applied to a wide variety of sample types. While traditional TMT methods are limited to comparing 6-11 samples per experiment due to tag availability, advanced TMTpro reagents now support up to 16-plex quantification, greatly increasing throughput and enabling more complex experimental designs.
2. Label-free Quantitative Proteomics
Label-free methods perform comparisons by measuring chromatographic peak areas/ion intensity ratios or counting MS2 spectra. Compared to stable isotope labeling methods, label-free approaches have lower reproducibility and accuracy, as all systematic and non-systematic variations can affect MS data. However, label-free methods offer several advantages. First, there is no limit to the number of samples that can be compared in an experiment. Second, label-free methods provide more efficient protein identification and quantification. Third, they offer a higher dynamic range for quantification compared to stable isotope labeling methods.
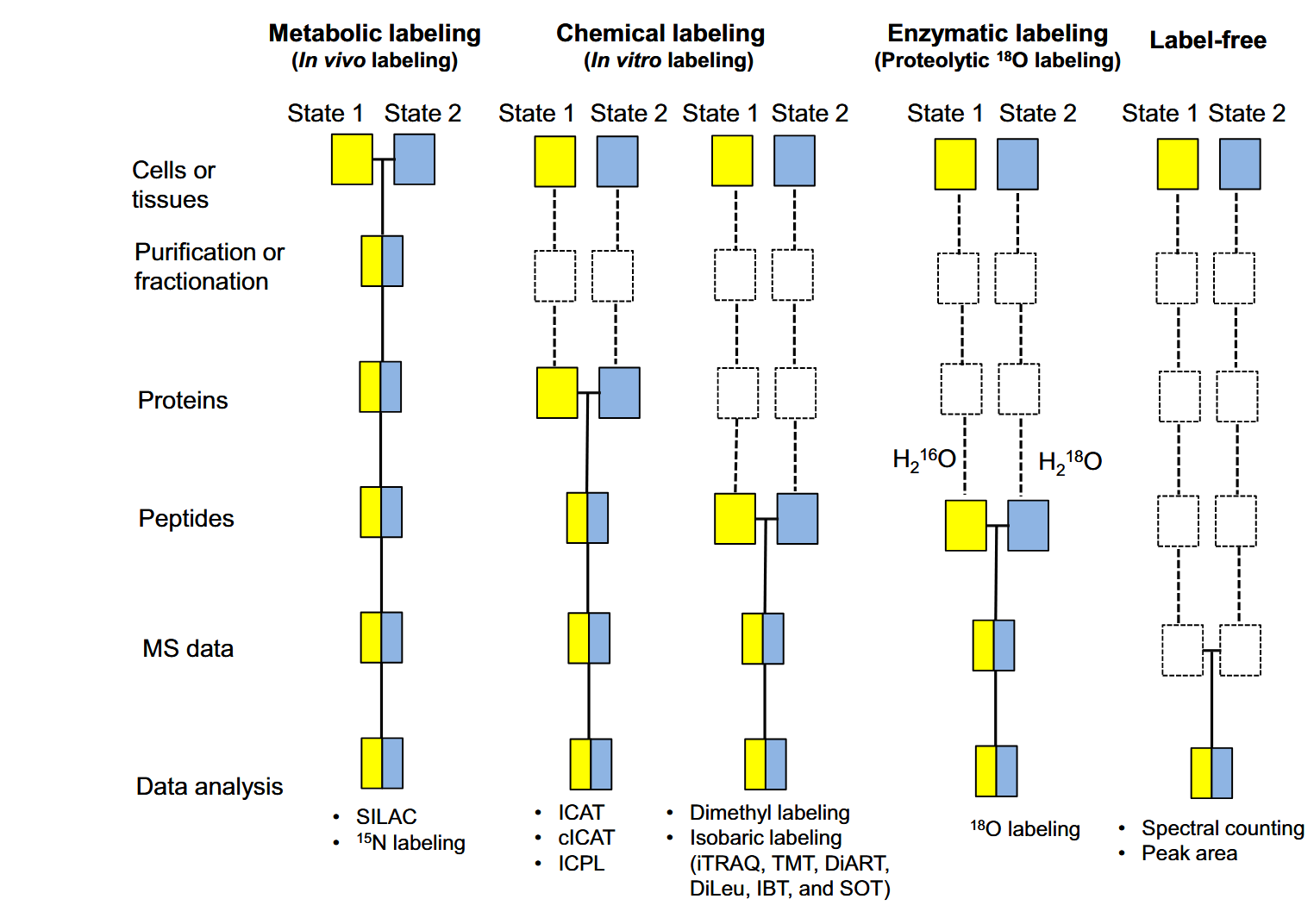
Chen, X. et al. Genomics Proteomics Bioinformatics. 2021.
Figure 1. MS-based Quantitative Proteomics Strategies
In addition to the above quantitative proteomics strategies, MtoZ Biolabs also offers other approaches such as iTRAQ and dimethyl labeling to meet various commercial and research needs. If you are interested in our services, please feel free to contact us.
Service Advantages
1. Advanced Mass Spectrometry Platform
MtoZ Biolabs is equipped with industry-leading mass spectrometry platforms, including Thermo Fisher Orbitrap series instruments (e.g., Orbitrap Fusion Lumos) combined with Nano-LC systems, providing high-sensitivity and high-resolution quantitative analysis to ensure data accuracy and reliability.
2. Broad Sample Compatibility
Our services accommodate a variety of sample types, including cells, tissues, plasma, and body fluids, enabling the analysis of complex biological samples and overcoming diverse experimental challenges.
3. Efficient Turnaround Time
Optimized workflows and efficient analytical platforms significantly reduce the experimental cycle, delivering precise results promptly to accelerate scientific progress.
4. Expert Research Team
Our team of experienced proteomics specialists excels in experimental design, sample processing, and data analysis, offering end-to-end solutions to fully support the achievement of your research objectives.
Applications
1. Quantitative Proteomics Reveals Disease Biomarkers
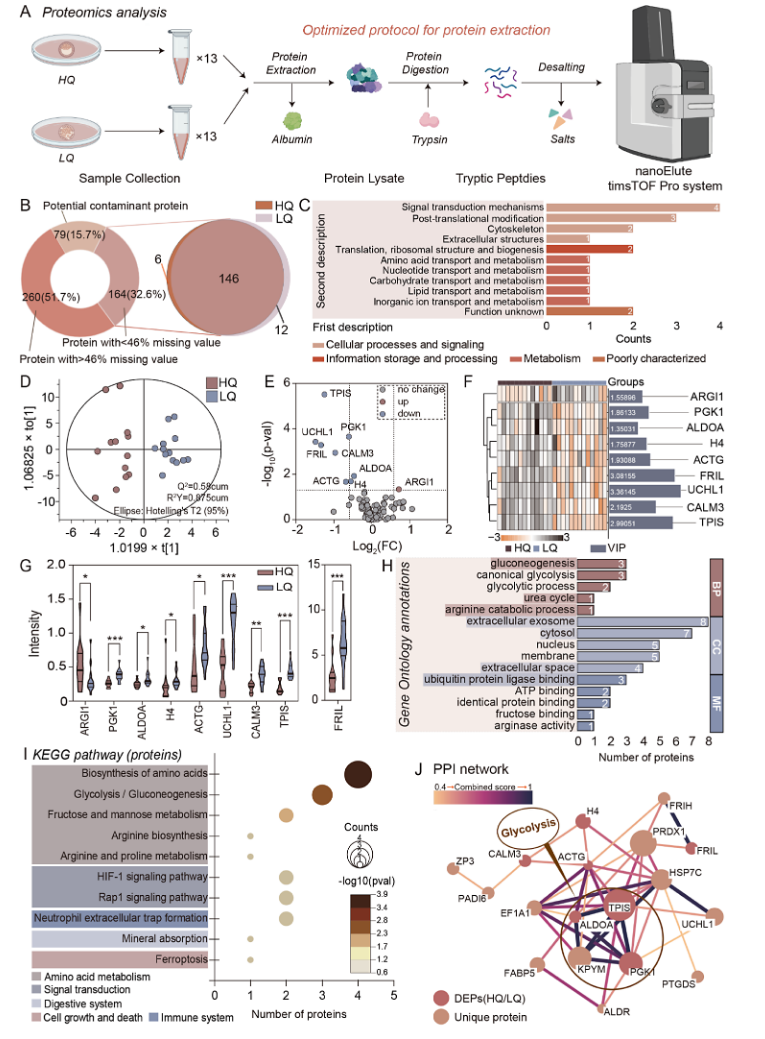
Deng, S. et al. Anal Chem. 2024.
2. Quantitative Proteomics Explores Therapeutic Mechanisms of Drugs
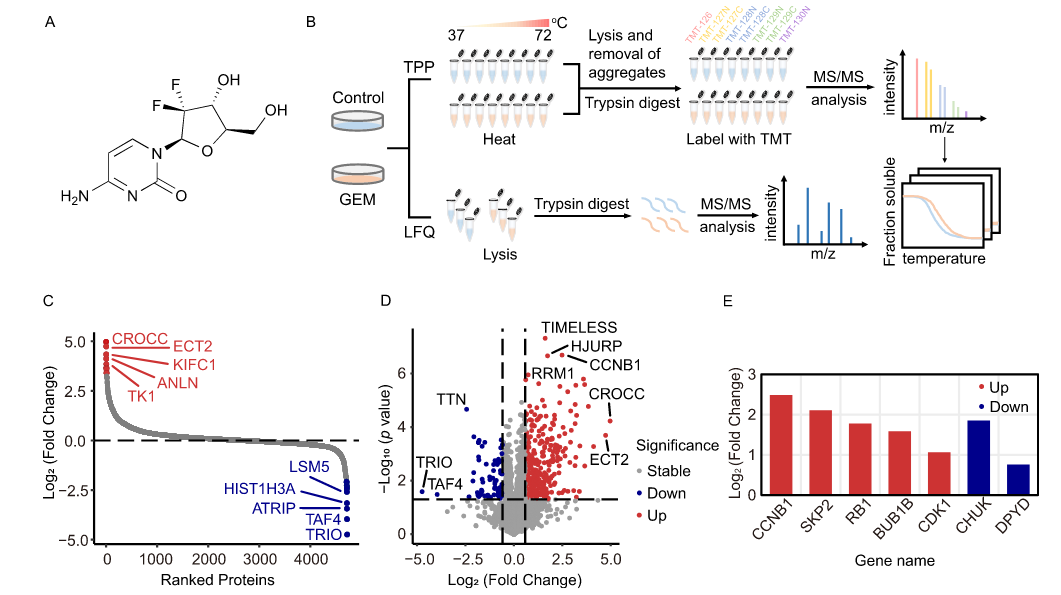
Jiang, Y. et al. J Proteome Res. 2024.
FAQ
1. Why can TMT only perform relative quantification and not absolute quantification?
TMT is an isotopic labeling method that enables relative quantification of multiple samples by labeling peptides with different isotopic tags. Its principle is based on comparing the signal intensities of labeled peptides across samples rather than measuring the absolute abundance of a specific peptide or protein.
Lack of Internal Standards: TMT-based quantification typically lacks absolute quantification internal standards or calibration curves, making it unable to directly measure the absolute abundance of proteins.
Instrument Response Inconsistency: The mass spectrometer's response factors (i.e., the instrument's response intensity to different peptides) can vary between proteins or peptides, preventing direct inference of absolute protein concentrations.
Deliverables
1. Comprehensive Experimental Details
2. Materials, Instruments, and Methods
3. Relevant Liquid Chromatography and Mass Spectrometry Parameters
4. The Detailed Information of Quantitative Proteomics
5. Mass Spectrometry Image
6. Raw Data
If you require our quantitative proteomics analysis service, welcome to learn more details!
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
Label-Based Protein Quantitative Service—iTRAQ, TMT, SILAC
How to order?

