





De Novo Sequencing Proteomics: When to Use It for Novel Peptides, Sequence Gaps, and Unexpected Variants
- informative spectra remain unassigned after a conventional database search
- the reference is incomplete, biologically mismatched, or too narrow
- the goal is novel peptide discovery, gap bridging, or variant hypothesis generation
- fragment ion series quality supports residue inference
- spectra are weak, chimeric, or heavily interfered
- an open search, error-tolerant search, or revised custom database is more likely to explain the data
- the claim depends on a unique full sequence call that bottom-up data may not support
- a full candidate peptide in favorable cases
- a sequence tag
- gap-supporting evidence
- a ranked list of candidate variants plus follow-up validation recommendations
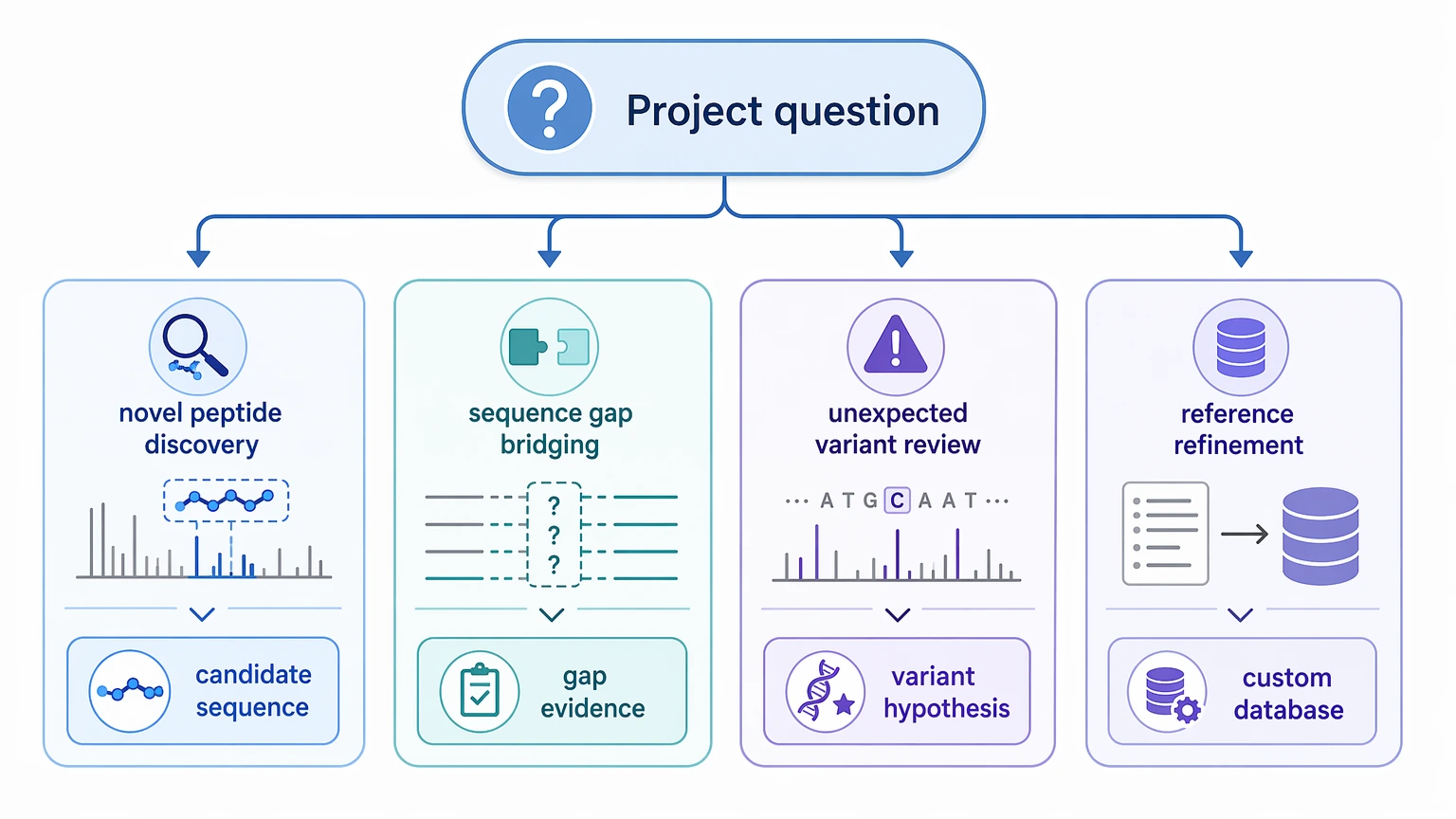
- Novel peptide discovery: recover candidate sequences or sequence tags for peptides absent from the current reference
- Sequence gap bridging: find evidence linked to a localized sequence gap in a partially known protein
- Unexpected variant review: test whether unexplained spectra support an amino acid substitution, splice variant, truncation, or unexpected processing product
- Reference refinement: generate sequence evidence that improves a later custom database rather than making a final biological claim
-
Look at the informative unassigned subset.
Low-intensity noise does not justify de novo. Clean, reproducible, unexplained spectra do. -
Review fragment ion series continuity.
Longer contiguous b ions / y ions support stronger residue inference than isolated fragments. -
Consider peptide length and composition.
Very short peptides may not be unique. Long, PTM-rich peptides may remain ambiguous. -
Ask whether the unknown is localized or diffuse.
A focused gap or a small cluster of variant-like spectra is a better use case than broadly unresolved proteome coverage. - a full candidate peptide sequence from selected spectra
- a longer sequence tag
- localized evidence for a sequence gap
- a ranked list of candidate unexpected variant interpretations
- annotations on confidence, ambiguity, and likely alternative explanations
- re-searching against a refined custom database
- checking reproducibility across independent runs or batches
- targeted confirmation by LC-MS/MS or MRM
- synthetic peptide comparison for top candidates
- orthogonal sequence confirmation when protein context is still unresolved
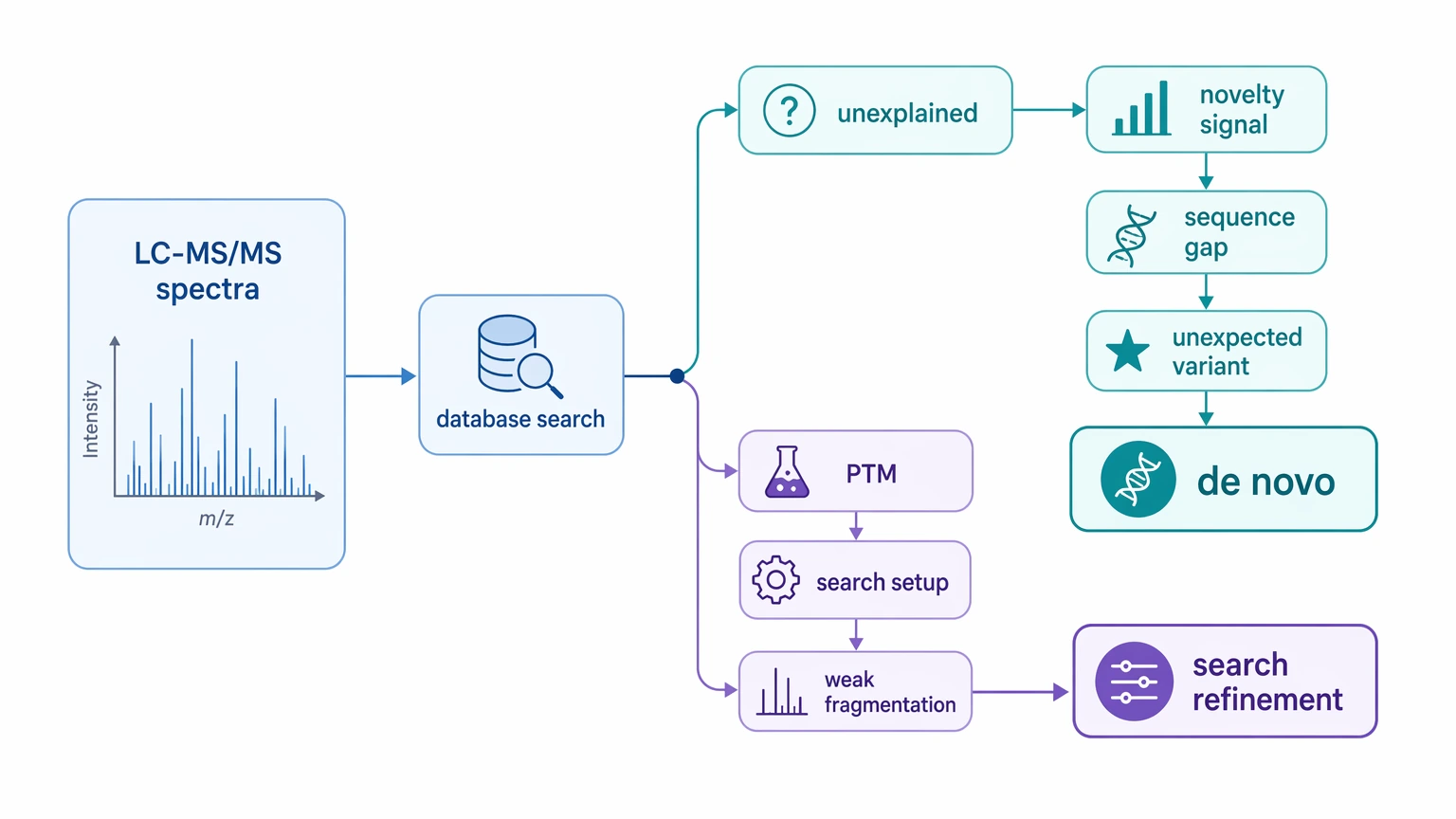
Add de novo sequencing proteomics when a well-tuned database search still leaves a meaningful set of high-quality LC-MS/MS spectra unexplained and the project has a real novelty signal, such as a non-model organism, an engineered construct, a localized sequence gap, or peptide evidence for an unexpected variant. If the underlying issue is poor search setup, missed post-translational modification (PTM) hypotheses, or weak MS/MS fragmentation, de novo interpretation is usually not the first step to escalate.
Quick Decision Guide
Use de novo sequencing proteomics when:
Do not start with de novo when:
Expected deliverables:
Where This Decision Usually Becomes Urgent
This decision usually comes up after an LC-MS/MS run looks technically solid but biologically incomplete. The dataset may show good precursor intensity, acceptable precursor mass accuracy, and many clean spectra, yet a notable fraction of informative features still lacks a confident peptide-spectrum match (PSM). In other cases, identified proteins explain most of the sample but not a missing stretch of sequence coverage, an unexpected truncation, or peptide evidence that fits an amino acid substitution, splice variant, or other proteoform missing from the reference.
At that stage, the team is not asking a broad method question anymore. It needs to decide whether the unknown really reflects missing sequence content or whether a better search model would recover the answer faster. That distinction matters. De novo work is most useful when it addresses genuine sequence novelty, not when it is used as a routine rescue step for every low-assignment dataset.
Why Standard Identification Fails in Novelty-Driven Projects
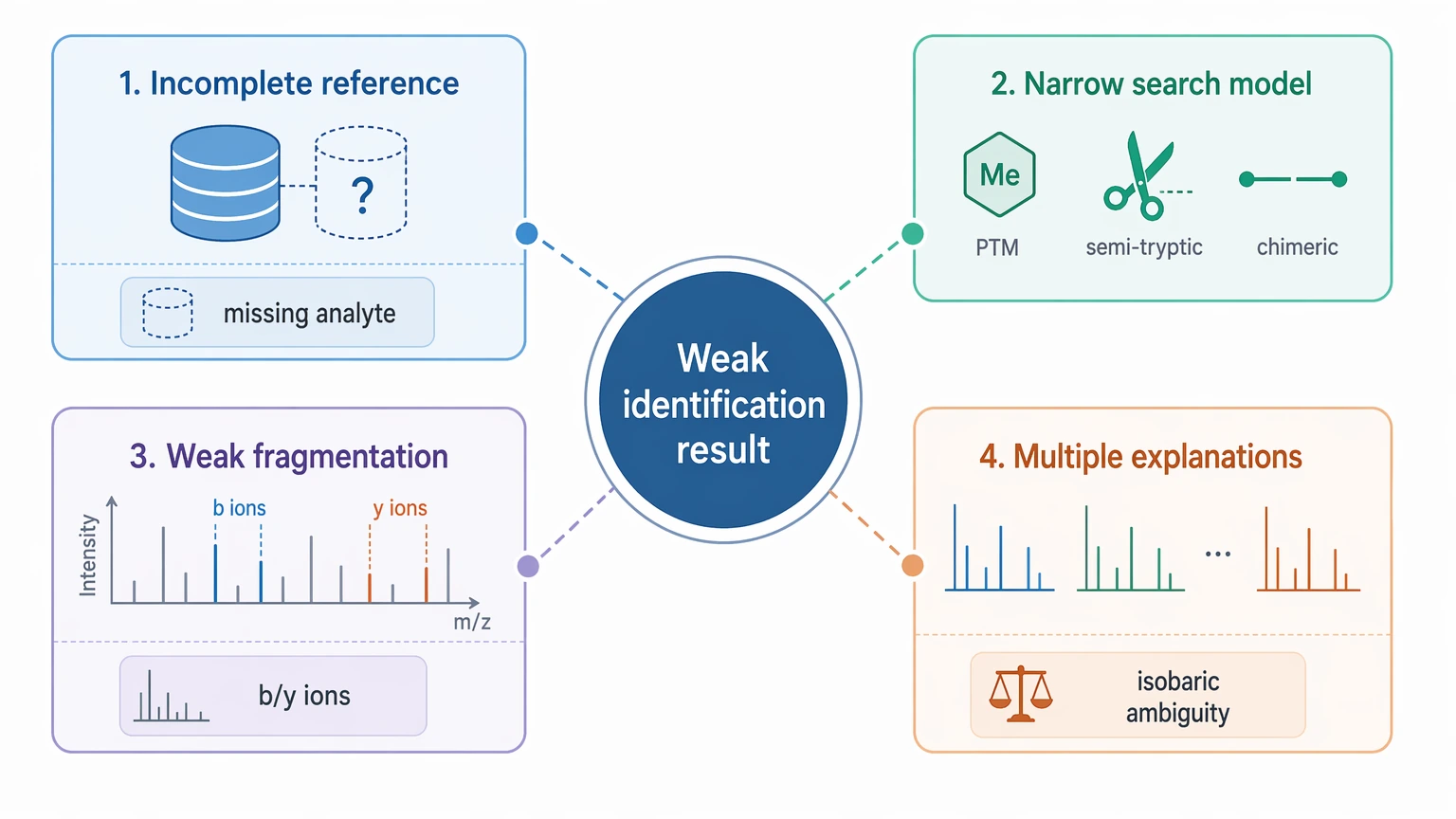
A weak identification result does not automatically justify de novo peptide sequencing. In this setting, four cause categories matter most.
1. The reference database does not represent the analyte
This is the clearest case for de novo interpretation. It shows up in non-model organisms, poorly annotated samples, engineered proteins, fusion constructs, venom peptides, heavily processed products, and impurity-related unknowns. If the analyte is not in the reference, repeating database matching will not recover it.
2. The search model is too narrow
Some unexplained spectra come from semi-tryptic peptides, unexpected cleavage products, unmodeled PTMs, or a reference that is correct in principle but not useful in practice. In those cases, an open search, error-tolerant search, or expanded custom database may resolve more spectra than de novo alone.
3. The spectra do not support confident residue inference
De novo sequencing proteomics relies on interpretable MS/MS fragmentation. Sparse b ions / y ions, short ion ladders, co-isolation, and spectral interference all reduce sequence confidence. Under those conditions, de novo output often narrows to a short sequence tag instead of a complete candidate peptide.
4. The biology allows more than one plausible explanation
PTM-rich peptides, mixed processing states, and isobaric substitutions can leave several reasonable interpretations on the table. A de novo result may still help, but it should be framed as a confidence-ranked proposal. This matters most when isobaric residue ambiguity, PTM placement, or database-search limits prevent a unique sequence call from tandem mass spectrometry alone.
Step 1: Define the Question Before Choosing the Method
Start by deciding what kind of answer the project actually needs. That choice shapes the workflow.
This keeps expectations grounded. Many projects do not need a full peptide call on the first pass. A well-supported tag or a short list of candidate sequences is often enough to guide follow-up searching and targeted confirmation.
Step 2: Check Whether the Data Are De Novo-Friendly
Before adding de novo interpretation, check whether the sample and spectra can support it.
The table below helps triage common sample contexts.
| Sample type | Best fit | Main constraint | Practical next step |
|---|---|---|---|
| Purified peptide or low-complexity fraction | Full candidate peptide calling | PTMs can still obscure residue order | Run de novo, then confirm top calls |
| Purified protein digest with a localized unknown region | Gap-bridging peptide or sequence tag | Bottom-up proteomics may not reconstruct the full protein | Pair de novo with database refinement |
| Engineered construct or fusion product | Junction or variant-localized evidence | Low-abundance junction peptides may be missed | Combine a construct-aware database with selective de novo |
| Non-model species digest | Novel peptide discovery in sparse annotation | Homology mapping may stay imperfect | Use de novo tags plus homology search |
| Highly complex whole-proteome digest | Selective follow-up on top unexplained spectra | Chimeric spectra reduce confidence | Triage first; do not apply blanket de novo |
Use the table as a screening guide, then confirm the fit against the available sample, spectra, and validation goal.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
Use four practical checks:
Step 3: Compare De Novo With Other Rescue Routes
De novo is most useful when it is chosen against real alternatives, not treated in isolation.
| Scenario | Recommended workflow | Key limitation | Follow-up need |
|---|---|---|---|
| Good spectra and likely missing reference content | Database search + de novo peptide sequencing | Some residues may remain ambiguous | Targeted confirmation of top candidates |
| Known sequence with suspected missed PTMs | Database search + open search | PTMs can mimic novelty | Confirm PTM localization |
| Engineered construct with expected design options | Custom database + error-tolerant search, then selective de novo | Search space may still miss unplanned products | Validate junction or variant peptides |
| Localized sequence gap in a known protein | Gap-focused de novo + refined search | Evidence may remain partial | Orthogonal validation or terminal confirmation |
| Broad low-assignment complex proteome | Reassess search setup and spectral quality first | Poor spectra limit every route | Repeat acquisition or simplify the sample |
Choose de novo when the unresolved issue is missing sequence content. If the main issue is search-space design, another route is often more direct.
If your team is at that point and needs a project-specific readout, MtoZ Biolabs can evaluate your project around unexplained LC-MS/MS spectra, reference limitations, and whether de novo interpretation or a revised search strategy is the more defensible next step.
Step 4: Match the Workflow to the Project Type
Novel peptides in database-limited studies
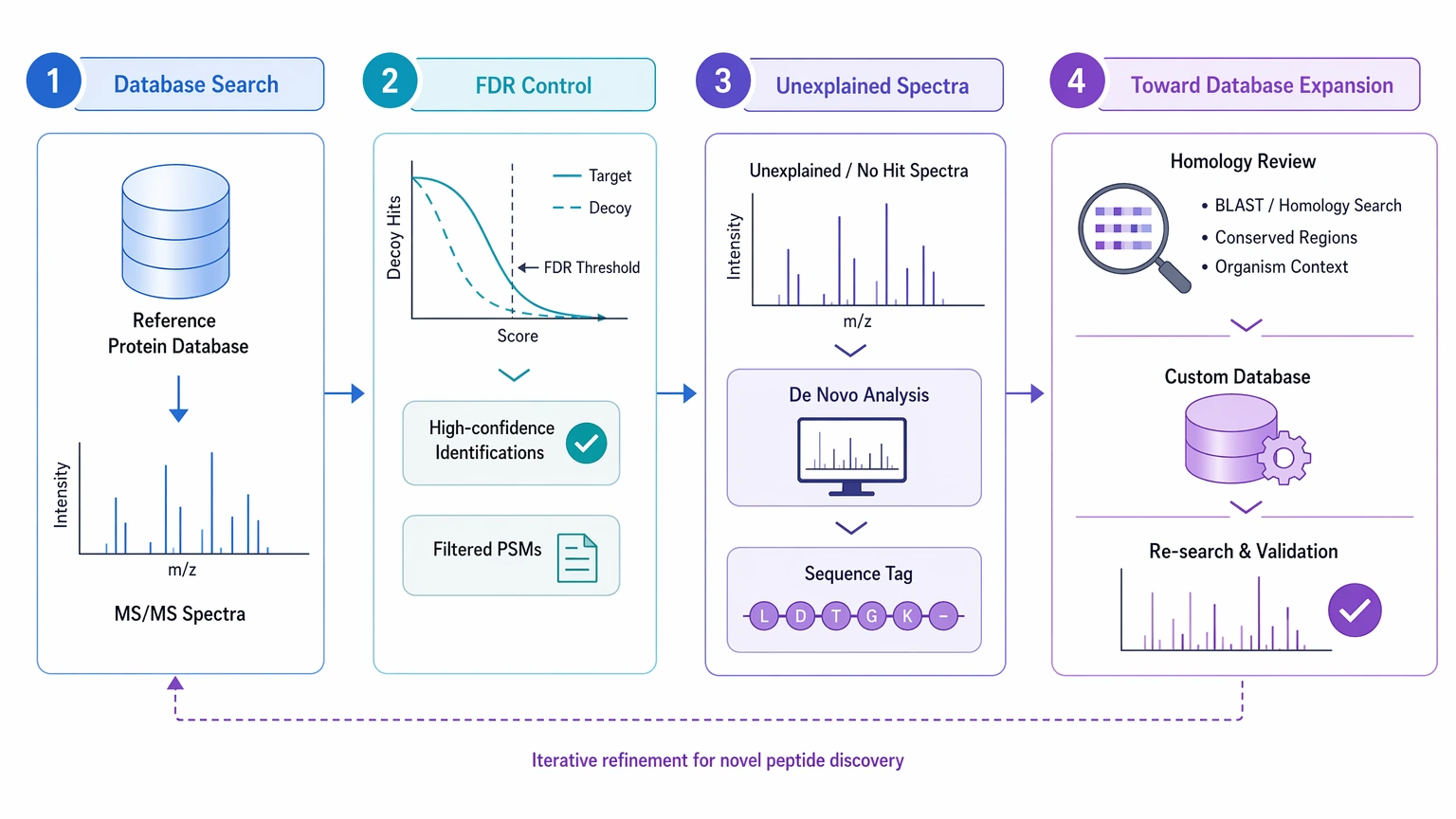
Run a standard database search first with appropriate false discovery control. Then send the highest-quality unexplained spectra to de novo analysis. Use the resulting sequence tag or candidate sequence list to support homology review or custom database expansion.
Sequence gaps in partially known proteins
Focus on peptides that flank or map near the missing region. The goal is not to rediscover the entire digest. It is to recover evidence that narrows or bridges the unresolved segment. When the missing region is terminal or structurally heterogeneous, another method may be more informative than repeated reinterpretation of bottom-up data.
Unexpected variants and altered processing
Treat de novo output as a variant hypothesis generator first. A candidate unexpected variant is most useful when fragment coverage localizes the change and competing explanations, especially PTMs, have already been tested. When leucine/isoleucine or modification ambiguity remains, the output should be reported as provisional rather than definitive.
Expected Results and Validation Methods
A good de novo decision does not mean every unknown turns into a unique sequence. It means the project gains interpretable evidence that moves the next decision forward.
Immediate deliverables may include:
Follow-up confirmation may include:
Keep the reporting boundary clear: de novo output can support a hypothesis, but biologically important claims usually need orthogonal validation before they are treated as confirmed sequence events.
Key Cautions and Practical Limits
Sample quality and amount limit what de novo can resolve
Low-input, degraded, or chemically heterogeneous material may produce identifiable signal but still fail to yield interpretable residue ladders. Keep enough sample in reserve for follow-up confirmation instead of using all material in discovery-only runs.
Controls and repeat observations matter
A novel-looking peptide is harder to trust when it appears once, in one batch, without supporting controls. Reproducibility across injections, preparations, or biological replicates lowers the chance that carryover or a transient contaminant is driving the call.
Batch effects and contamination can imitate novelty
Keratin, digestion artifacts, and cross-sample carryover can generate unmatched or misleading spectra. Review contamination patterns before turning unexplained signals into a novelty claim.
Interpretation boundaries are real
De novo interpretation from tandem mass spectrometry can leave residue positions unresolved, especially in PTM-rich peptides or when isobaric residue ambiguity is present. Bottom-up evidence may support a sequence model or a shortlist, but it does not always provide full protein certainty.
Another method may be the better next step
If spectra remain weak, the unresolved region is too broad, or intact structural context is central to the question, a different route may be more efficient. That may mean improved acquisition, sample fractionation, terminal sequencing, top-down confirmation, or outside bioinformatics support instead of more de novo analysis.
Conclusion
De novo sequencing proteomics is most useful when standard identification has already done the obvious work and the remaining unexplained evidence still points to genuine missing sequence content. The best candidates are projects with clean LC-MS/MS data, plausible database limitations, localized sequence gaps, or spectra that credibly support an unexpected variant without stretching the claim too far. In those settings, the practical output is often a candidate peptide, a sequence tag, or a ranked variant model that directs the next validation step.
For non-model species studies, engineered proteins, purified peptide samples, and datasets with a persistent block of informative unassigned spectra, a combined workflow is often easier to defend than repeated database searching alone. If that matches your project stage, contact MtoZ Biolabs to submit your requirements and discuss the sample context, unresolved spectra, and the most suitable confirmation path before you commit more sample or budget.
FAQ
Can de novo sequencing proteomics help when the reference database is mostly correct but still incomplete?
Yes. That is a common middle-ground use case. A mostly correct reference may still miss splice junctions, engineered regions, truncations, or rare processing products. In that setting, de novo output often works best as sequence evidence for refining the database rather than replacing it.
What is the minimum useful outcome if full peptide calling fails?
A partial sequence tag can still matter if it narrows homology, points to a gap-adjacent region, or supports inclusion of candidate peptides in a revised search space. Its value comes from whether the tag changes the next experimental decision.
How should teams interpret a single strong unexplained spectrum?
Treat it cautiously. One strong spectrum can justify follow-up review, but not a broad biological conclusion on its own. Reproducibility, contamination review, and an alternative-search check should come before a novelty claim.
Is de novo helpful for heavily modified peptides?
Sometimes, but only with caution. PTMs can obscure residue order, shift fragment patterns, and expand the list of plausible interpretations. In PTM-rich samples, de novo is often more useful for generating constrained candidates than for claiming a unique final sequence.
When does homology search add more value than another de novo pass?
Homology search becomes especially useful when de novo produces informative but incomplete tags from organisms or proteins with sparse annotation. A partial tag may still map to a conserved family or domain even when full peptide reconstruction is not possible.
What should a team prepare before requesting a workflow review?
Bring the project objective, sample type, digestion strategy, search settings already used, the pattern of unassigned spectra, examples of key MS/MS scans, and any prior open search or error-tolerant search results. That makes it easier to decide whether the next step is de novo interpretation, database refinement, or targeted confirmation.
How to order?

