





De Novo Protein Sequencing by Mass Spectrometry: When It Fits Unknown Protein and Peptide Characterization Projects
- Choose de novo protein sequencing or de novo peptide sequencing when the database search gives no match, only low-score partial matches, or conflicting assignments.
- Use it when the target may contain a substitution, truncation, cyclization, or unexpected terminal change.
- Use it when the project needs residue-level confidence or a sequence tag rather than simple mass agreement.
- Prioritize it only when the precursor ion and fragment ions are clean enough to support spectral annotation.
- Choose a hybrid or alternative workflow first when peptide mapping against a known reference can answer the question.
- Switch to another route when intact mass analysis is enough to confirm mass shifts or heterogeneity, or when co-isolation and formulation background dominate the spectra.
- a candidate sequence for a short peptide
- a sequence tag for a larger analyte
- localization of a sequence variant within a digest
- PTM-aware interpretation with ambiguity notes
- residue-level confidence by region rather than a single global call
Mass spectrometry-based de novo sequencing is the right first-line route when the true sequence is unknown or poorly documented, a standard database search cannot recover a credible match, and the LC-MS/MS data are clean enough for database-independent sequence inference. In practical terms, it is most useful for unknown peptide identification, sequence variant follow-up, impurity-related peptide analysis, and partially documented proteins.
It is a weaker starting point when the sample is highly mixed, tandem mass spectrometry coverage is thin, or a reference-based method can answer the decision question more directly. The best way to triage the project is to define the required deliverable first, then check whether the analyte class, sample condition, and fragment-ion evidence support residue-level interpretation.
Quick decision guide
Why teams switch from database search to de novo interpretation
Most groups do not begin with de novo protein sequencing. They move to it when database search stops answering the actual project question. The sequence may be missing from the database, a substitution or truncation may break search assumptions, a PTM may weaken matching, or the team may need a direct read on an unknown peptide instead of a broad identification label.
That distinction matters in unknown protein characterization. A weak database hit does not automatically justify a full de novo workflow. Sometimes the spectra are too poor, the precursor ion is contaminated, or the underlying question is better handled by peptide mapping or targeted confirmation. The decision should be driven by sequence novelty and spectral interpretability, not by search failure alone.
When de novo sequencing is a realistic fit
The clearest fit is usually an unknown therapeutic peptide or another short analyte with one dominant species and a readable fragmentation pattern. In that setting, de novo peptide sequencing can generate candidate sequences, sequence tags, and local confidence calls that support development decisions.
A second fit is a partially documented recombinant protein. In practice, de novo protein sequencing here usually means a bottom-up or middle-down strategy that reconstructs informative regions, checks termini, and resolves missing or inconsistent records across multiple peptides.
A third fit is a sequence variant case. If a digest contains a suspected substitution or truncation that standard database search does not place cleanly, hybrid interpretation is often the better route. Database search can anchor the known regions, while de novo sequence inference clarifies the altered stretch.
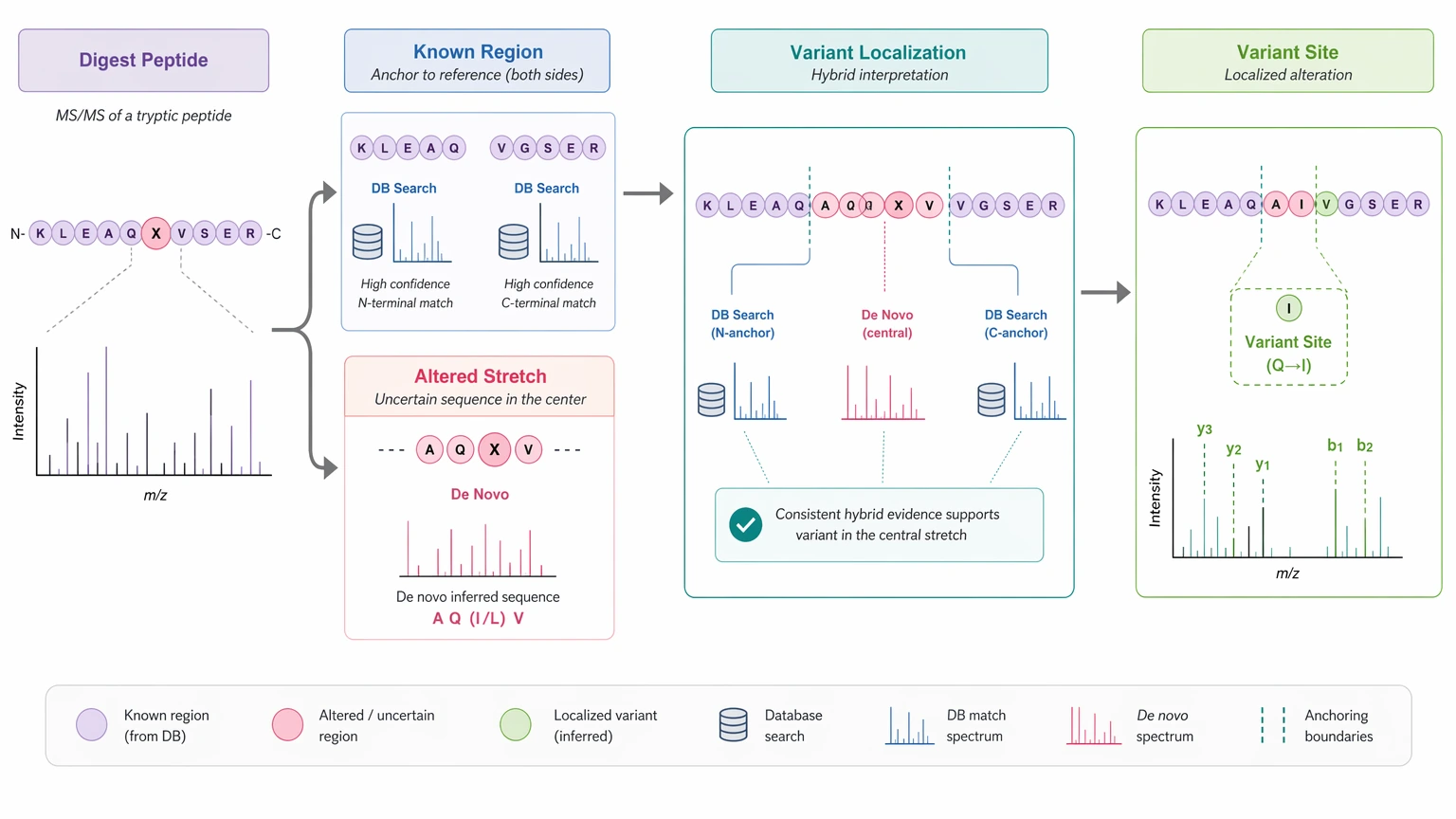
A fourth fit is an impurity-related peptide project. If one impurity yields a distinct precursor ion and interpretable fragment ions, de novo analysis can help determine whether the component is a degradation product, a truncated form, or another unexpected peptide species. PTM-rich analytes can also be workable when the goal is to rank candidates and map uncertainty rather than force a single final call.
A method-selection workflow for project triage
1. Define the exact output before choosing the method
Start with the end point. If the team only needs to confirm a known backbone, peptide mapping plus database search is usually faster than de novo protein sequencing. If the team needs unknown peptide identification, a ranked candidate sequence list, or sequence-tag evidence for an undocumented region, de novo becomes more reasonable.
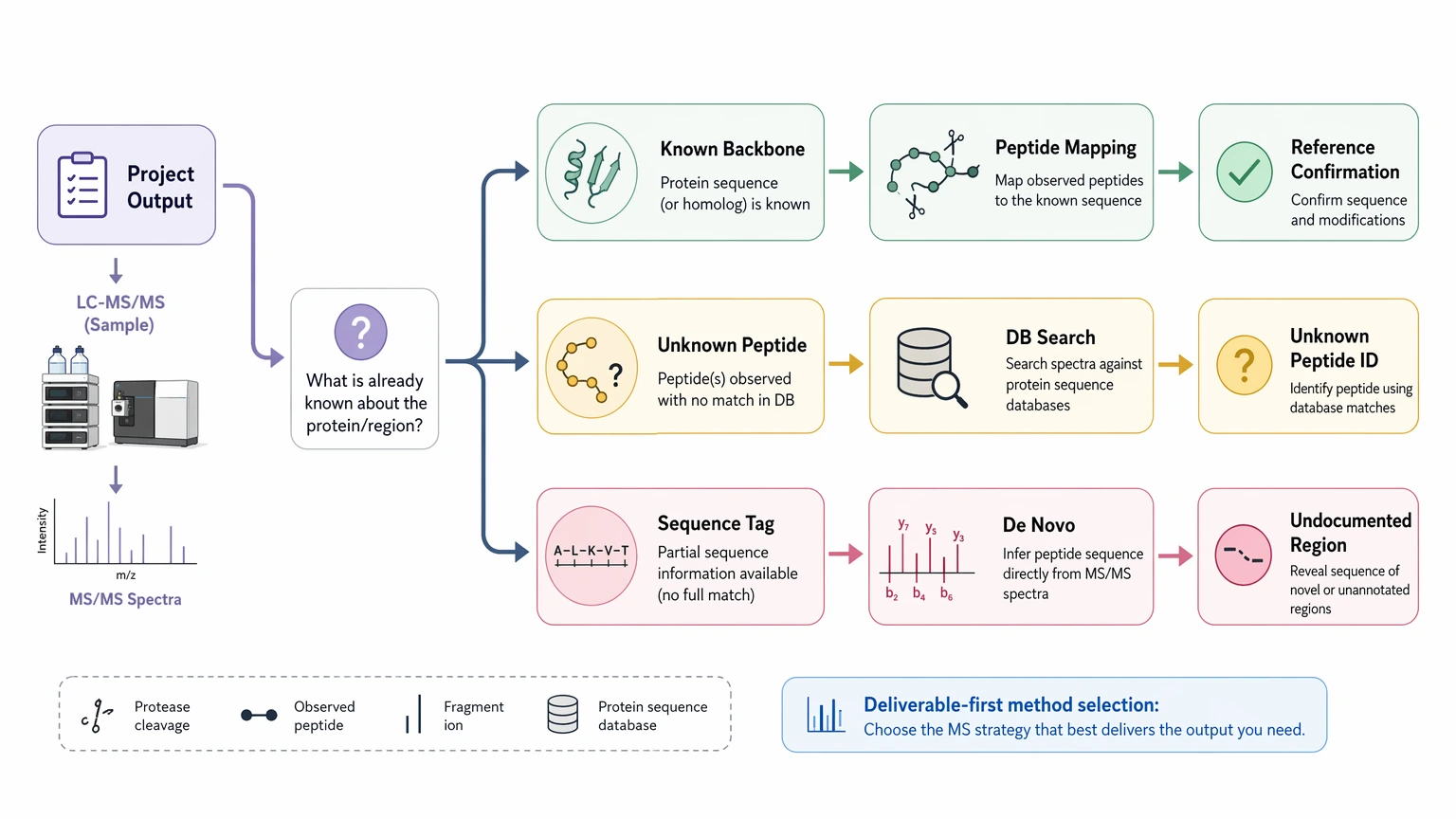
2. Check whether the analyte class supports residue-level interpretation
Short peptides and defined protein-derived peptides are usually easier to interpret than large heterogeneous proteins, so analyte class should be checked before scarce material is committed to extended sequencing work.
| Analyte class | Typical fit | Why it fits | Main constraint |
|---|---|---|---|
| Unknown peptide | High | Cleaner LC-MS/MS and simpler sequence space | Terminal blocking or cyclization may hide ions |
| Impurity-related peptide | Moderate to high | Distinct precursor ion may allow direct interpretation | Mixed impurity pools can create chimeric spectra |
| Sequence variant in digest | Moderate | Hybrid analysis can localize altered residues | Low-abundance variant signals may limit confidence |
| Larger undocumented protein | Moderate | Multi-digest workflows can recover informative regions | Full reconstruction is labor-intensive and incomplete in some regions |
| PTM-rich analyte | Case-dependent | Complementary fragmentation may preserve modification information | PTMs can suppress or redirect fragmentation |
3. Review sample and LC-MS/MS evidence before committing material
If the best spectra do not support a credible manual review in the key regions, software scoring alone will not rescue the project. Repeat injections, cleaner precursor isolation, or another analytical route may be more useful than forcing a de novo readout from weak evidence.
| Decision metric | Favorable sign | Warning sign |
|---|---|---|
| Sample composition | One dominant species | Multiple co-eluting components |
| Sample amount | Enough for repeat LC-MS/MS and follow-up confirmation | Only one limited injection remains |
| Precursor ion isolation | Clean precursor ion and defined charge state | Co-isolation or unstable charge distribution |
| Fragmentation pattern | Continuous ion series | Large gaps, dominant neutral loss, or weak sequence coverage |
| Mass accuracy | Consistent fragment assignments | Variable mass error or uncertain annotation |
| PTM burden | Limited or interpretable PTMs | Multiple labile or blocking modifications |
4. Choose the right interpretation model
Use de novo as the main route when the sequence is truly absent from the database and the fragmentation pattern supports direct inference. Use a hybrid model when peptide mapping, intact mass analysis, or database search can narrow the candidate space while de novo interpretation addresses the unknown region. Choose another method first when the real need is reference confirmation, presence-absence testing, or mixture screening rather than sequence reconstruction.
If your team has raw files but limited time for fragmentation strategy, spectral annotation, or candidate ranking, you can submit your requirements and evaluate your project with MtoZ Biolabs before committing scarce material to a full sequencing workflow.
When de novo sequencing is not the best first choice
De novo sequencing is not the default answer to every unclear identification. It is usually a weak first step for highly mixed samples, severe formulation background, intact-only protein data without supporting digestion, or projects where the biological question does not require residue order. It also becomes less efficient when a known reference sequence already exists and the main goal is confirmation rather than discovery.
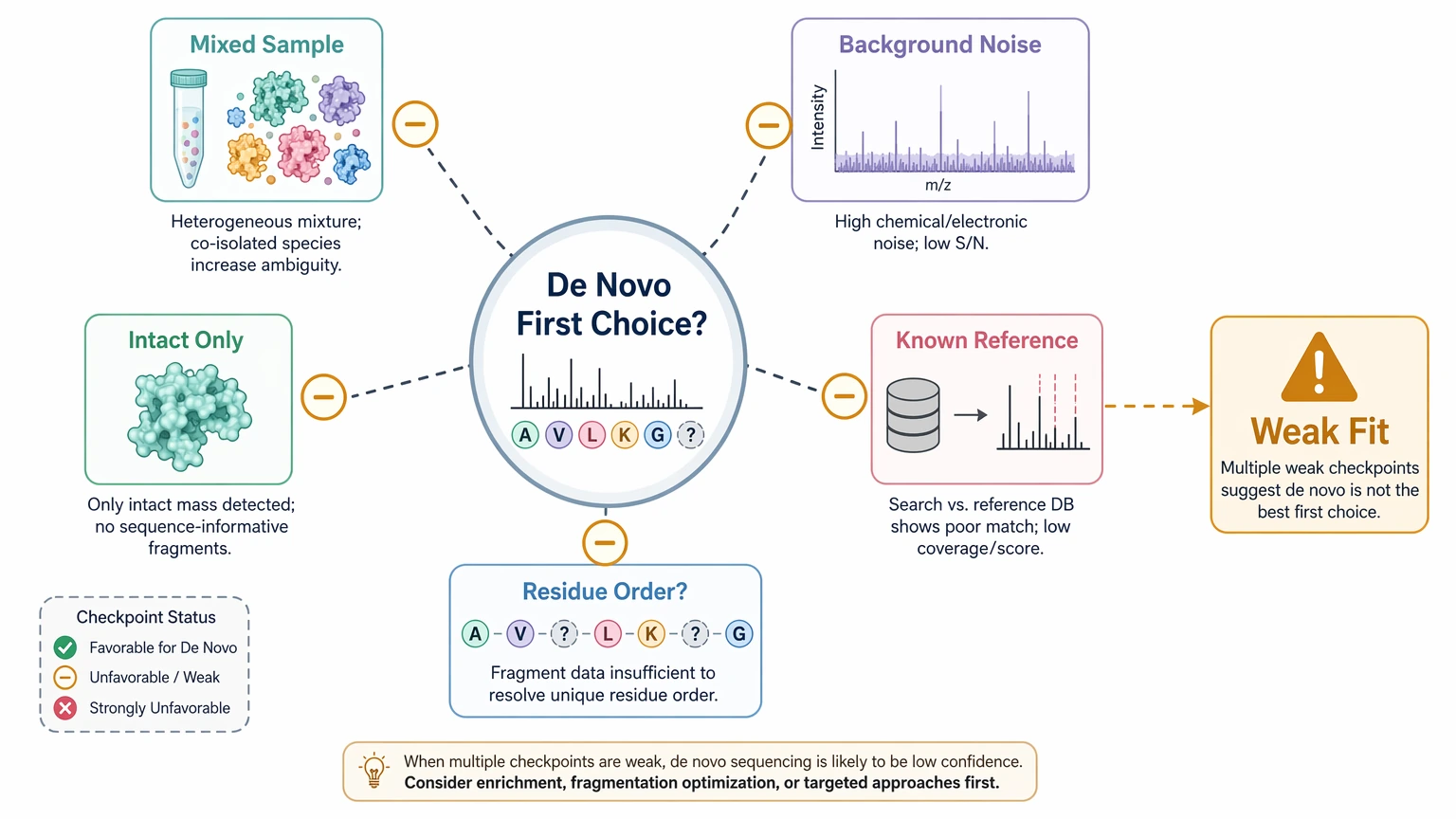
There are also hard interpretation limits. Standard LC-MS/MS often cannot reliably distinguish isobaric residues such as leucine and isoleucine, and PTM localization may remain local rather than absolute when coverage is incomplete. Fragment-ion gaps, co-isolation, and PTM complexity can leave uncertainty even in otherwise useful datasets.
Expected results and validation methods
Teams often expect a final sequence string, but the first deliverable is often narrower and more defensible: ranked candidate sequences, annotated spectra, sequence tags for uncertain regions, PTM interpretation where supported, residue-level confidence mapping, and a clear list of unresolved positions.
Follow-up confirmation should be planned early. Depending on the project, that may include repeat LC-MS/MS runs to confirm the same fragmentation pattern, alternative fragmentation using HCD, CID, ETD, or EThcD, consistency checks against intact mass analysis, peptide synthesis comparison for short candidates, and targeted validation of termini or variant sites.
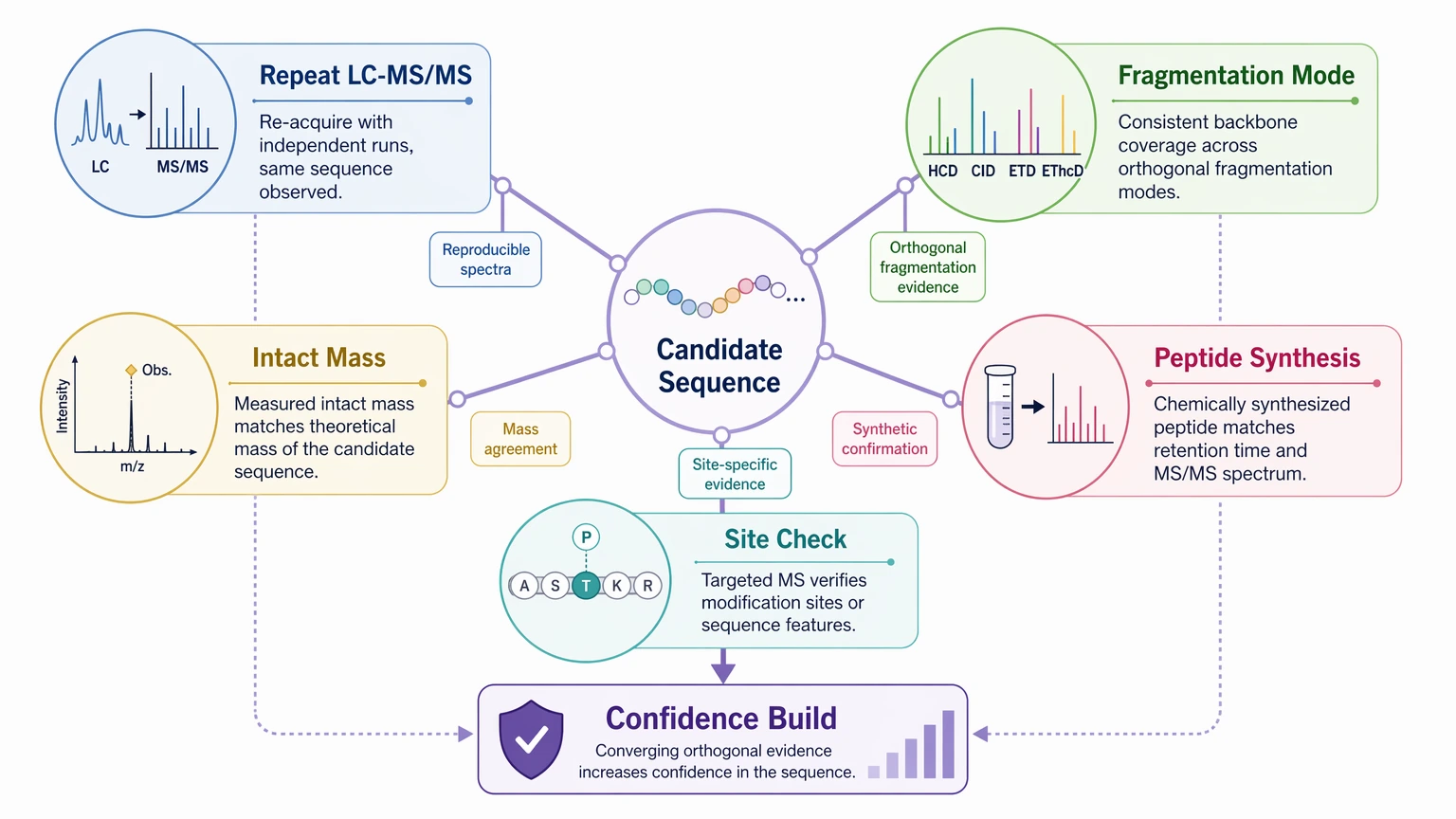
For outsourcing decisions, ask for both interpretation and a confirmation plan. In complex unknown protein characterization or impurity-related peptide work, MtoZ Biolabs can review existing LC-MS/MS data, define the likely confidence level, and help you submit your requirements for a project that includes both sequence interpretation and orthogonal validation planning.
Service Routes to Consider
For projects that need sequence reconstruction, confirmation of uncertain residues, or a practical validation path, the following routes usually map well to the decision point:
Key cautions and practical limits
Sample amount and sample quality should be checked before launch. If little material remains, the project may produce only a sequence hypothesis rather than a confirmable answer, because repeat analysis and alternate fragmentation usually consume additional sample.
Controls and repeat design still matter. A candidate sequence is more persuasive when key fragment ions reproduce across runs or preparations, and variant or impurity cases benefit from comparison with the closest known reference or process context.
Batch effects and contamination can distort precursor selection and create chimeric spectra. Salts, excipients, co-eluting species, and carryover may reduce interpretability, and cleanup can improve spectra but also consume material. Data interpretation also has boundaries: sequence confidence is often local rather than uniform across the full analyte, and unresolved isobaric residues or PTM-related gaps may remain even after careful review.
If the sample stays highly mixed, if the main question is reference confirmation, or if intact mass and peptide mapping already answer the decision point, a de novo workflow may add cost without adding enough clarity. In that situation, targeted LC-MS/MS, peptide mapping, or intact mass analysis is often the better next step.
Conclusion
De novo protein sequencing by mass spectrometry is most justified when a real sequence gap remains after database search and the LC-MS/MS data contain interpretable fragment-ion evidence for database-independent sequence inference. It is especially relevant for unknown peptides, sequence variant follow-up, impurity-related peptide analysis, and partially documented proteins, while mixed samples, sparse MS/MS data, and purely confirmatory questions usually point to other methods first.
If you are working with limited material, PTM-rich analytes, or uncertain sequence confidence, the most practical next step is to define the required deliverable, align the validation path, and contact us to discuss the project context, sample constraints, and existing data with MtoZ Biolabs before the next round of analysis.
FAQ
Can de novo sequencing still help if the sequence is partly known?
Yes. Known regions can anchor interpretation, while de novo analysis focuses on the uncertain stretch, terminal change, or sequence variant.
What files are most useful for a project assessment?
Raw LC-MS/MS files are best, along with search results, precursor masses, digest information, formulation notes, and a short statement of the decision the sequence result must support.
Does a no-hit database search automatically justify de novo sequencing?
No. First check whether the failure came from the search setup rather than true sequence novelty. A narrow database, missing variable modifications, the wrong enzyme setting, poor precursor isolation, or low-fragmentation quality can all produce a no-hit result. De novo work is more defensible when you still have a clean precursor, a readable ion series, and enough material for repeat LC-MS/MS or an orthogonal confirmation run.
Is intact mass analysis enough for undocumented proteins?
Usually not by itself. Intact mass can tell you whether the observed mass is plausible, whether multiple forms are present, and whether a major truncation or modification may exist, but it cannot usually place residue order or localize most sequence changes. It becomes more useful when paired with subunit analysis, peptide mapping, or targeted MS/MS on suspicious regions, especially if the protein is heterogeneous or carries PTMs.
What is a realistic success target for a larger protein project?
A practical target is often to answer the project decision, not to force full sequence closure. For a larger protein, success may mean confirming termini, covering the regions most likely to contain a variant, or generating high-confidence sequence tags that narrow the candidate space. To improve the odds, prepare for multiple digests, repeat injections, and a plan for how unresolved leucine/isoleucine positions or modified sites will be handled in follow-up confirmation.
When should a team outsource instead of handling interpretation internally?
Outsourcing becomes reasonable when the project needs multiple fragmentation modes, careful ambiguity reporting, candidate ranking for unknown peptide identification, or a validation plan beyond standard database search output.
Related Services
How to order?

