





Why Antibody Sequencing Is Difficult: Common Failure Points in Sequence Recovery Projects
- starting material type: purified antibody, hybridoma, supernatant, or mixed biological source
- storage and handling history: freeze-thaw exposure, age, and aggregation risk
- monoclonal purity: whether a single antibody species is still the dominant component
- available quantity: whether enough material remains for orthogonal validation
- unresolved chain pairing
- incomplete CDR evidence
- leucine/isoleucine ambiguity at decision-critical positions
- mixed clone signals from a drifting hybridoma
- missing orthogonal validation for the intended use case
- What is the starting material? Protein, cells, supernatant, and mixed fractions each come with different information limits.
- What is the real deliverable? Partial characterization and expression-ready sequence are not interchangeable.
- Is monoclonal purity established? Functional performance alone does not prove sequence homogeneity.
- Can orthogonal validation be reserved? A small sample may not support both recovery and confirmation.
- Which uncertainty would actually block the project? Chain pairing, CDR ambiguity, or clone heterogeneity do not carry the same weight in every program.
Antibody sequence recovery usually becomes difficult for four concrete reasons: the starting material no longer carries enough unique information, heavy-chain and light-chain evidence cannot be paired with enough confidence, the variable region, especially the complementarity-determining regions (CDRs), is not covered evenly, or the final dataset still contains residue-level uncertainty that blocks recombinant re-expression. So when a project fails, it is not just a generic protein sequencing problem. In most stalled programs, the real issue is a mismatch between the sample source, the sequence target, and the level of certainty required for downstream use.
A protein-only workflow may recover useful candidate sequences, but that still does not guarantee an expression-ready result. A hybridoma- or transcript-based route can improve variable heavy chain (VH) and variable light chain (VL) recovery, yet uncertainty may remain when clonal drift or mixed populations are present. The practical question is not whether any sequence can be reported. It is whether the reported sequence is specific enough for construct design, clone rescue, or sequence confirmation.
What Antibody Sequencing Actually Needs to Deliver
Antibody sequencing can mean amino acid sequence recovery from protein material or nucleic acid sequence recovery from cells or transcripts. In rescue projects, though, the target is usually narrower and harder to satisfy: paired VH and VL information complete enough to support recombinant re-expression, engineering, or redevelopment of an assay reagent.
That distinction matters because partial analytical recovery and expression readiness are not the same endpoint. A report may show strong peptide evidence across framework regions (FRs), yet still leave uncertainty in CDR3, chain pairing, or leucine/isoleucine identity. For project teams, those unresolved details often decide whether the result is usable or still exploratory.
Why Antibodies Are Harder to Sequence Than Many Proteins
Antibodies are repetitive in some regions and highly diverse in others. The FRs often resemble known antibody patterns, while the CDRs contain the sequence features most closely tied to binding specificity. That uneven distribution of information makes antibody sequencing harder than routine protein identification, where conserved database matching may be enough.
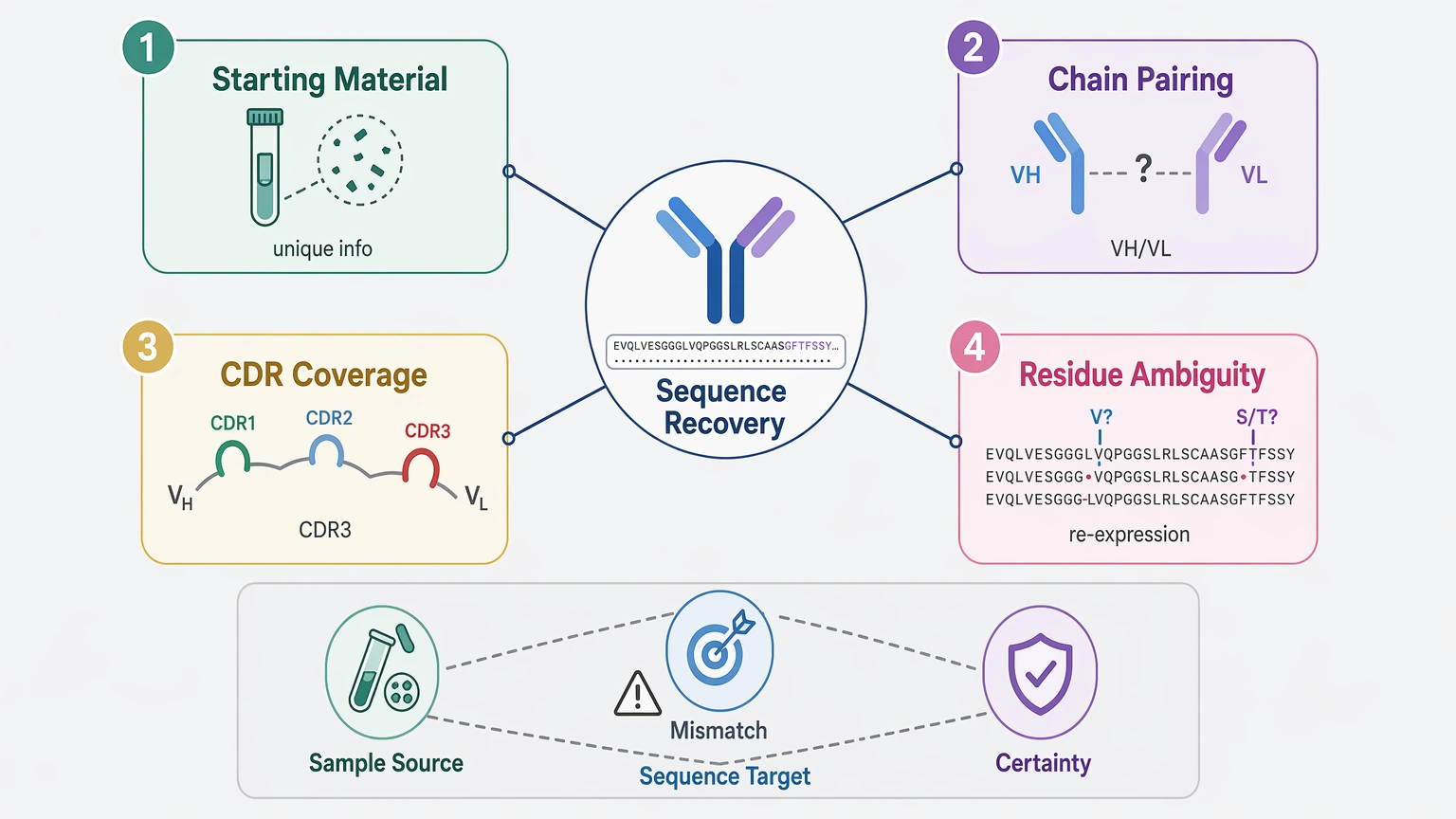
Antibodies also create a two-chain reconstruction problem. Heavy chain and light chain are separate polypeptides linked by disulfide bond architecture, but purified protein does not preserve clone-linked pairing information in a form that peptide data can always read directly. If more than one antibody species is present, even a technically sound de novo sequencing workflow can return several plausible VH or VL candidates.
A third complication is that antibody projects often begin with compromised historical materials: aged hybridoma lines, purified monoclonal antibody with incomplete records, low-quantity samples, or supernatants with uncertain monoclonal purity. In those settings, the sequencing challenge is partly molecular and partly archival.
The Most Relevant Failure Points in Sequence Recovery Projects
1. Starting Material Sets the Ceiling for Sequence Recovery
The first limitation appears before LC-MS/MS, RACE, or next-generation sequencing (NGS) data are even interpreted. If the sample is degraded, aggregated, limited in amount, or contaminated with additional antibody species, the project starts with less sequence uniqueness than expected.
Protein-only material is especially restrictive because it lacks direct transcript linkage. Serum-derived fractions and impure supernatants can look acceptable in routine protein assays while still carrying substantial background complexity. Aged hybridoma material brings a different risk: the cells may no longer represent the original productive clone.
Before starting another sequencing round, teams should confirm four practical points:
If those basics are unclear, the next experiment may reproduce the same ambiguity instead of resolving it.
2. Chain Pairing Uncertainty Is a Core Protein-Based Limitation
When the source is purified antibody protein, detected peptides can often be assigned to heavy chain or light chain, but secure chain pairing is a separate issue. Put simply, the workflow may recover plausible VH and VL sequences without proving which pair actually belongs together.
This matters most when the goal is recombinant re-expression. A candidate heavy chain paired with the wrong light chain may still look reasonable on paper, yet fail after expression. That is why protein-derived sequence recovery should be judged against the intended deliverable, not just against overall peptide yield.
If your next decision depends on whether protein-only data can support expression design, submit your requirements early and evaluate the project against the actual need for chain pairing rather than generic sequence coverage.
3. CDR Coverage Gaps Create Disproportionate Risk
Not all missing residues matter equally. FR gaps can sometimes be resolved through homology context or follow-up evidence. CDR gaps are more disruptive because they sit in the most functionally informative part of the variable region and usually allow less inference.
The pattern below explains why headline coverage percentages can mislead:
| Region | Common recovery issue | Project impact |
|---|---|---|
| FRs | Higher homology and easier peptide assignment | Often useful for scaffold confirmation, less decisive for specificity |
| CDR1/CDR2 | Short peptides or uneven digestion products | May allow more than one sequence path |
| CDR3 | Highest diversity and sparse confirming evidence | Often the main bottleneck for expression-ready sequence calls |
| Chain termini | N-terminal blockage or poor recovery of end peptides | Can interrupt variable-region assembly |
For antibody sequencing, peptide coverage distribution matters at least as much as total peptide coverage. A report with strong FR recovery and weak CDR3 evidence may still be analytically useful, but it should not be treated as expression-ready by default.
4. MS-Based De Novo Sequencing Has Real Residue-Level Limits
LC-MS/MS is central to protein-based antibody sequencing, but the spectra do not always support one unambiguous sequence solution. The best-known example is leucine/isoleucine ambiguity, since those residues are isobaric. Standard fragmentation evidence often cannot separate them directly without added context.
Post-translational modification (PTM) burden adds another layer of difficulty. Glycosylation, oxidation, deamidation, and pyroglutamate-related N-terminal blockage can alter mass patterns and complicate peptide mapping. Disulfide bond architecture also affects digestion behavior and peptide recovery. So a sequence model may be well supported in broad outline while still containing unresolved local choices.
That does not make LC-MS/MS uninformative. It means the output has to be interpreted at the right confidence level: candidate sequence set, high-confidence variable-region recovery, or expression-ready sequence after orthogonal validation.
When RACE or NGS Changes the Risk Profile
RACE and NGS become especially valuable when clone-linked cellular material is still available. They can improve access to VH and VL transcript information, reduce dependence on peptide-only reconstruction, and help fill variable-region gaps that are hard to resolve from protein data alone.
Even so, transcript recovery is not a universal fix. Hybridoma instability, nonproductive transcripts, and mixed populations can still produce more than one plausible answer. In those cases, nucleic acid evidence may explain why the project remains ambiguous, but it does not automatically restore a unique clone identity.
A combined workflow is often most useful when the team needs to reconcile what the cells encode with what the antibody protein actually contains.
Why a Reported Sequence May Still Not Be Safe for Re-Expression
Researchers often treat “sequence recovered” as the finish line, but sequence recovery and recombinant re-expression readiness are different thresholds. A result may still be unsuitable for construct design if any of the following remain:
This is where many projects stall. A sequencing report can be technically solid and still fall short of the exact downstream action the team needs.
In projects built around missing clone records, uncertain purified antibody stocks, or ambiguous hybridoma materials, MtoZ Biolabs can evaluate the sample and workflow fit before you submit your requirements for another sequencing attempt.
Pre-Project Checks That Reduce Avoidable Ambiguity
Before restarting a failed effort, clarify the points below with the sequencing team:
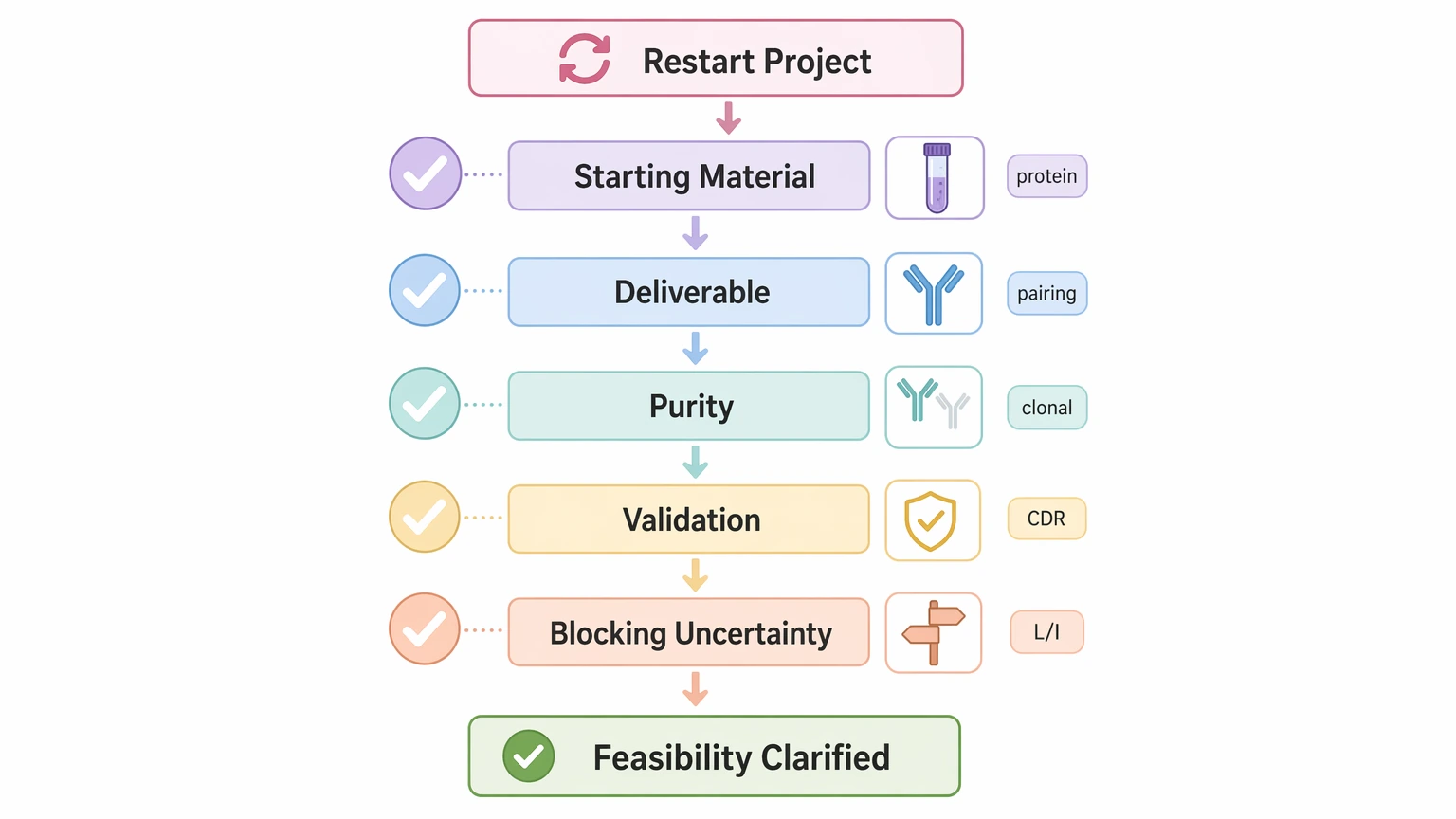
These questions help narrow the failure mode before more material and time are committed.
Conclusion
Antibody sequencing is difficult because the information that matters most to the project usually sits in the regions that are hardest to reconstruct, and many rescue efforts begin with samples that no longer preserve complete clone identity. In practice, the main question is whether the available material can support unique variable-region recovery, credible chain pairing, and enough CDR confidence for the intended downstream use. For teams working with hybridoma material, purified monoclonal antibody, or incomplete historical records, the most useful next step is often a bounded feasibility review that defines likely outputs, remaining ambiguity, and suitable validation paths. If that matches your situation, contact MtoZ Biolabs to evaluate your project and discuss what evidence would be sufficient for sequence interpretation, construct design, or a follow-up recovery strategy.
FAQ
Can constant-region information compensate for weak variable-region recovery?
Usually not. Constant-region peptides can confirm antibody class or support general identity, but they do not resolve the variable-region details needed for binding-specific interpretation or recombinant re-expression.
Why do two sequencing attempts on the same antibody sometimes disagree?
The disagreement may reflect real sample heterogeneity, differences in digestion coverage, or different interpretation thresholds for ambiguous residues. It does not always mean one dataset is wrong.
Is purified monoclonal antibody ever enough without cellular material?
Sometimes yes, especially for characterization-focused goals. It is less reliable when the project requires unique VH/VL pairing and high-confidence CDR resolution for expression construct design.
What makes hybridoma-derived projects unexpectedly difficult?
An old hybridoma may no longer behave like the original clone. Clonal drift, mixed populations, or nonproductive transcripts can all complicate sequence recovery even when cells are still available.
Should a team repeat the same workflow after an ambiguous result?
Not automatically. First determine whether the previous ambiguity came from sample-source limits, chain pairing uncertainty, CDR coverage gaps, or residue-level interpretation limits. Repeating the same workflow without changing that constraint may not improve the outcome.
How to order?

