





When De Novo Protein Sequencing Is the Right Choice for Unknown Proteins or Database-Mismatched Results
- De novo protein sequencing is most useful after peptide mapping and database search fail to explain repeated, interpretable LC-MS/MS evidence.
- It is not the default response to every database-mismatched result; wrong database content, narrow post-translational modification (PTM) settings, low spectral quality, or mixed samples may still be the main issue.
- De novo peptide sequencing and protein sequence reconstruction are related but not interchangeable. Strong peptide sequence tags do not always support a complete protein-level conclusion.
- Immediate outputs often include partial sequence reconstruction, sequence variant evidence, truncation evidence, or PTM-aware interpretation rather than a fully resolved sequence.
- Orthogonal validation usually determines whether a de novo result becomes decision-useful for unknown protein identification or biologics characterization.
- unmatched peptides recur across repeats
- MS/MS fragmentation supports interpretable peptide sequence tags
- the reference sequence cannot absorb the mismatch
- the project needs sequence-resolving evidence, not another best-match score
- the sample may contain a novel protein, undocumented sequence, truncation, fusion boundary, or clustered sequence variant
- database composition is incomplete or outdated
- PTM search settings are too narrow
- fragment ions are sparse or inconsistent
- the sample is visibly mixed, contaminated, or too weak for confident interpretation
- de novo peptide sequencing outputs for unmatched peptides
- peptide sequence tag assignments with fragment-ion support
- partial protein sequence reconstruction across informative regions
- evidence for sequence variant clusters, truncation, or altered N-terminus / C-terminus processing
- confidence assignment notes for ambiguous positions, gaps, and PTM-affected regions
- intact mass comparison against the proposed interpretation
- targeted LC-MS/MS confirmation of key peptides
- orthogonal digestion to test sequence continuity
- transcript or construct cross-checking where sequence records exist
- terminal analysis when N-terminus or C-terminus claims are central
When peptide mapping and routine database search still leave credible LC-MS/MS evidence unexplained, the real issue is not whether de novo protein sequencing is "advanced." The issue is whether the remaining mismatch comes from a search setup that can still be fixed or from a true sequence problem that needs direct inference. De novo protein sequencing makes sense when the project has reached the point where tandem mass spectrometry evidence must speak for itself because routine matching no longer explains what the sample is showing.
Key Takeaways
Quick Decision Guide
Escalate to de novo protein sequencing when:
Do not escalate first when:
What De Novo Protein Sequencing Adds Beyond Database Matching
A database search asks which known reference sequence best fits the spectra. De novo protein sequencing asks what amino acid order the spectra actually support, even when the right reference sequence is missing or wrong.
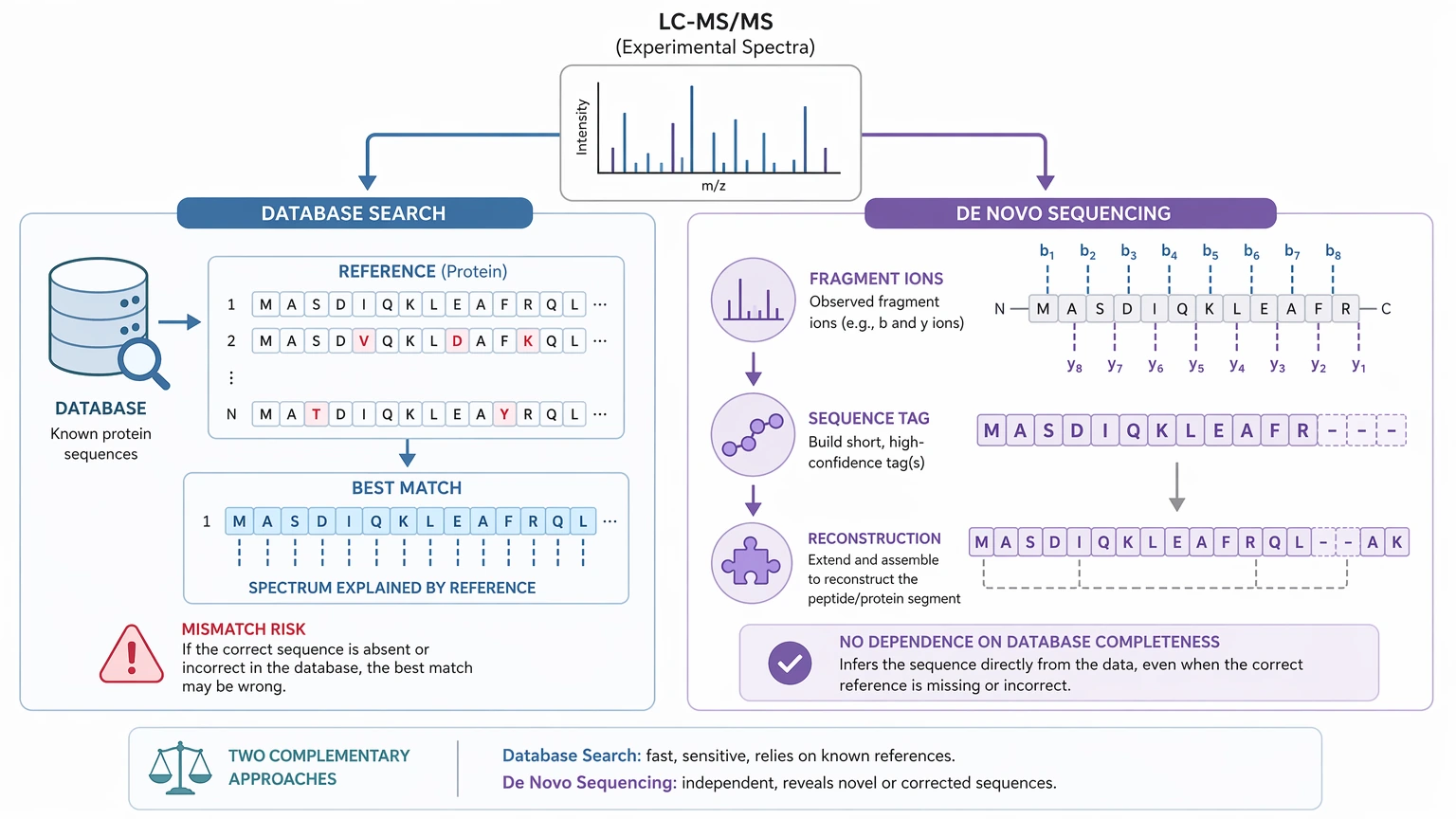
At the peptide level, de novo peptide sequencing uses MS/MS fragmentation to infer residue order from fragment ions, especially b ions and y ions. At the protein level, those peptide sequence tags still have to be assembled into a protein sequence reconstruction, a localized mismatch model, or a bounded-confidence assignment for specific regions.
That gap matters when database-mismatched results are under review. A project can produce several convincing peptide tags and still stop short of a full N-terminus-to-C-terminus reconstruction. Confidence often drops in PTM-rich regions, terminal regions, poorly fragmented peptides, or positions affected by isobaric residue ambiguity such as leucine and isoleucine. In practical terms, MS/MS evidence may support a strong local sequence while still leaving some residue identities or peptide connections uncertain at the whole-protein level.
When Re-analysis Still Comes Before De Novo Sequencing
Before escalating, look at the failure modes that most often look like true sequence mismatch in this setting.
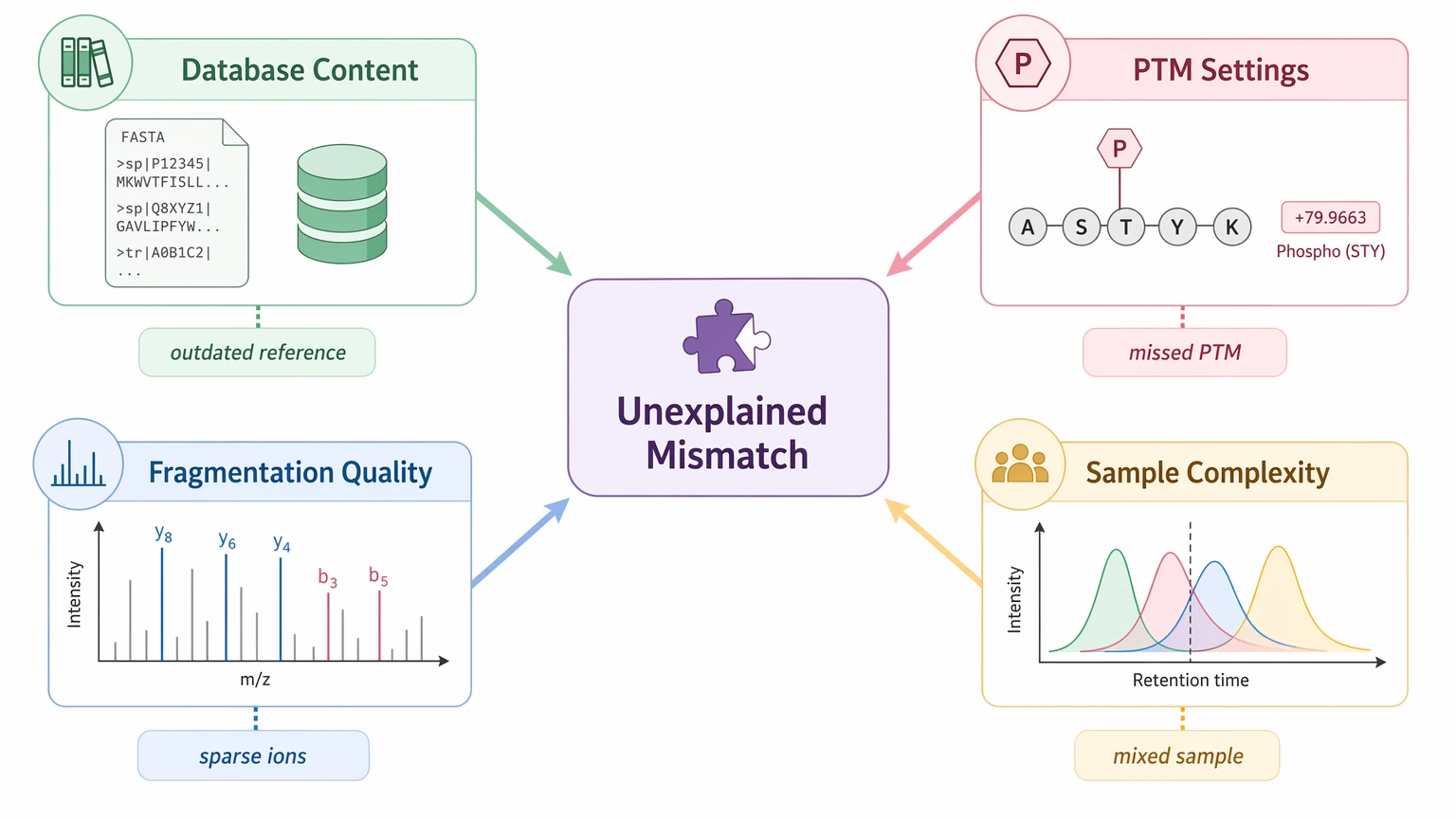
Database composition does not reflect the real sample
A search can miss the correct answer when the reference sequence is incomplete, outdated, or missing construct-specific features such as signal peptide processing, fusion regions, host-cell proteins, or lot history. This comes up often with proprietary constructs, legacy biologics, and poorly documented materials.
PTM handling is too narrow
A peptide can appear sequence-mismatched when the real issue is an unmodeled PTM. Oxidation, deamidation, glycation, clipping, and other modifications can shift precursor mass or alter fragment interpretation enough to suppress an otherwise valid database match.
MS/MS fragmentation is not informative enough
De novo interpretation needs rich, continuous fragmentation. If fragment ions are sparse, precursor isolation is poor, or signal is weak, a no-hit result may simply mean the spectra do not yet support a confident call.
Sample complexity is masking the target
Mixed proteins, background species, or co-eluting contaminants can create conflicting peptide assignments. In those cases, cleaner isolation, alternative digestion, or repeat acquisition may add more confidence than moving too early into a de novo workflow.
When De Novo Protein Sequencing Is the Right Choice
The best cases usually look the same in one respect: database-mismatched results remain after targeted re-analysis, and the unresolved evidence affects an actual project decision.
1. The protein source is undocumented or weakly referenced
If the sample comes from a non-model source, historical material, custom construct, or uncertain biologic lot, the reference sequence may not be reliable enough to anchor interpretation. In that setting, unknown protein identification often requires sequence inference rather than a database-dependent label.
2. Unmatched evidence is reproducible and localized
Repeated no-hit peptides, low-score clusters, or fragment-ion ladders that consistently disagree with the expected sequence mean far more than one unexplained spectrum. This pattern often points to a true sequence variant, truncation, or undocumented sequence segment.
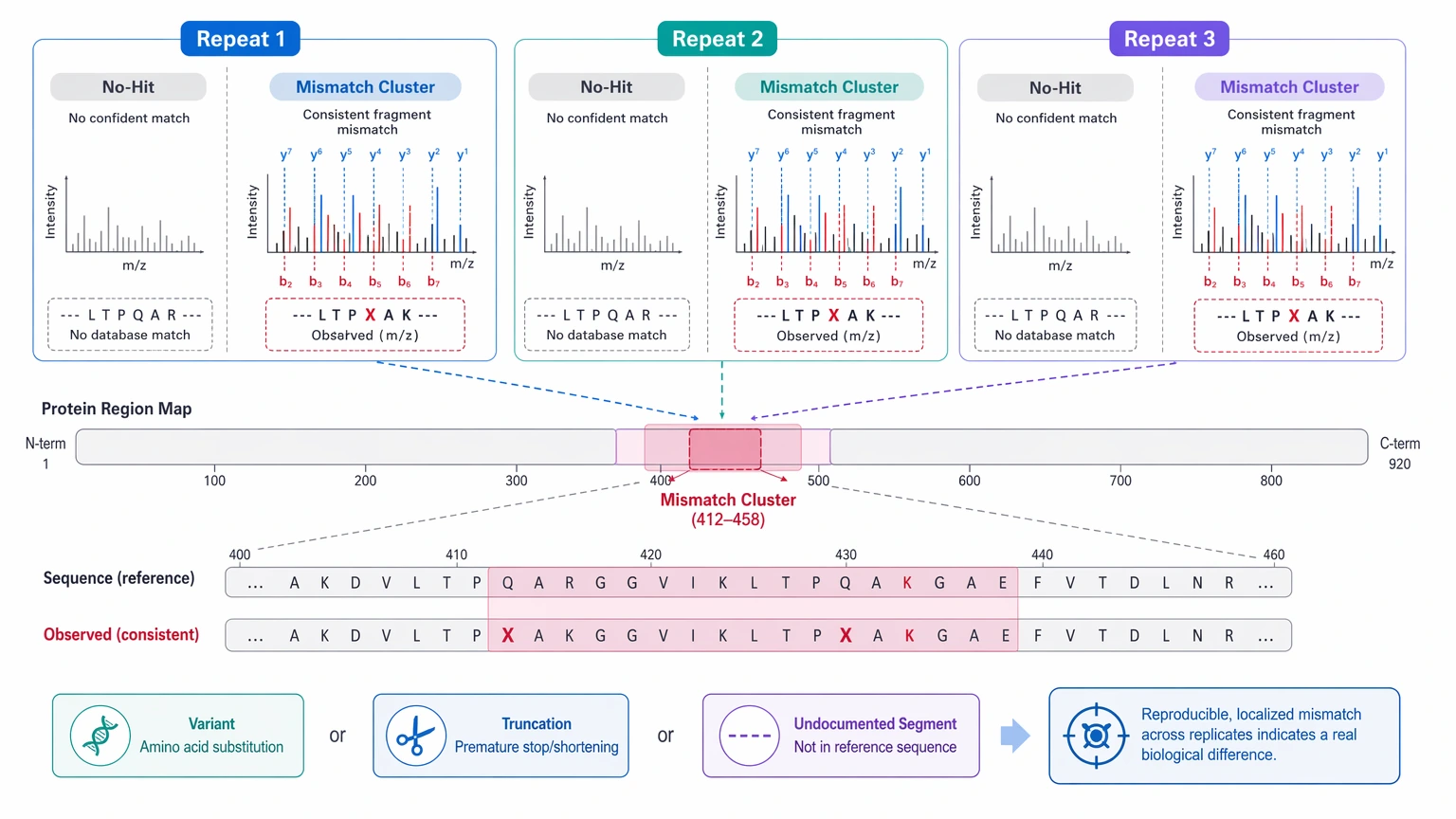
3. The mismatch changes the biological or product interpretation
If the unresolved region affects construct identity, processing state, fusion boundaries, or release-relevant characterization logic, peptide tags alone may not be enough. The project needs protein sequence reconstruction, or at least a defensible localized sequence model.
4. The sample supports peptide-to-protein assembly
De novo sequencing becomes easier to justify when sequence coverage is reasonably distributed, unmatched peptides are not isolated singletons, and intact mass or terminal evidence helps connect the peptide-level findings.
If your team is deciding whether the evidence has crossed that threshold, submit your requirements early so the workflow can be scoped around sample amount, purity, digestion strategy, and the decision the data must support.
Evidence Checklist Before You Commit
Use the table below to judge whether the case is ready for de novo protein sequencing or still needs another round of troubleshooting.
| Evidence | What it supports | Main limitation | Next step |
|---|---|---|---|
| Reproducible unmatched peptides with rich MS/MS fragmentation | De novo peptide sequencing and local sequence inference | Protein-level assembly may stay partial | Confirm the same region across repeats |
| Clustered mismatch within one region of an expected protein | Local sequence variant or truncation analysis | Does not justify full-length claims by itself | Add region-focused follow-up |
| Intact mass offset from the reference sequence | Global mismatch or altered processing state | Intact mass does not define residue order | Pair with peptide-level evidence |
| Clear N-terminus or C-terminus anomaly | Truncation, clipping, or processing difference | Terminal coverage may remain incomplete | Use terminal-focused confirmation |
| High purity and simple composition | Better protein sequence reconstruction | Purity alone does not remove ambiguity | Consider orthogonal digestion |
Takeaway: the strongest cases combine interpretable spectra, repeated mismatch behavior, and a real need for sequence-level resolution.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
Expected Results and Validation Methods
A realistic de novo workflow should separate immediate deliverables from follow-up confirmation.
Immediate deliverables may include:
Follow-up confirmation may include:
For teams deciding between re-analysis, de novo peptide sequencing, and protein-level reconstruction, MtoZ Biolabs can evaluate your project and help you submit your requirements with the right sample history, LC-MS/MS evidence, and validation goal.
Key Cautions and Practical Limits
De novo protein sequencing can clarify a hard mismatch, but the decision only makes sense if its limits are part of the plan.
Sample quality or amount limits: low abundance, degraded protein, or insufficient material can restrict digestion options and reduce sequence coverage.
Controls and repeat expectations: a single unexplained spectrum is weak evidence. Repeat acquisition, technical consistency, and comparison with expected material usually matter more than one high-scoring tag.
Batch or contamination risk: mixed lots, carryover, host-cell background, and co-isolated peptides can distort protein sequence reconstruction and make novelty look stronger than it is.
Interpretation boundaries: de novo peptide sequencing does not automatically prove a full undocumented protein. PTMs, database-search limits, and MS/MS interpretation uncertainty can leave unresolved gaps or preserve alternative explanations.
When another method is the better next step: if the main question is bulk identity confirmation, contaminant screening, or PTM profiling rather than sequence reconstruction, refined database search, intact mass analysis, or targeted validation may be more efficient than escalating to full de novo work.
How to Choose the Next Step
Use the project decision, not the novelty of the method, as your guide.
If the sample has a credible reference sequence and the mismatch is narrow, start with database search review, PTM expansion, and targeted confirmation. If the source is undocumented, the expected construct is uncertain, or the mismatch region changes the identity claim, de novo protein sequencing becomes the better path.
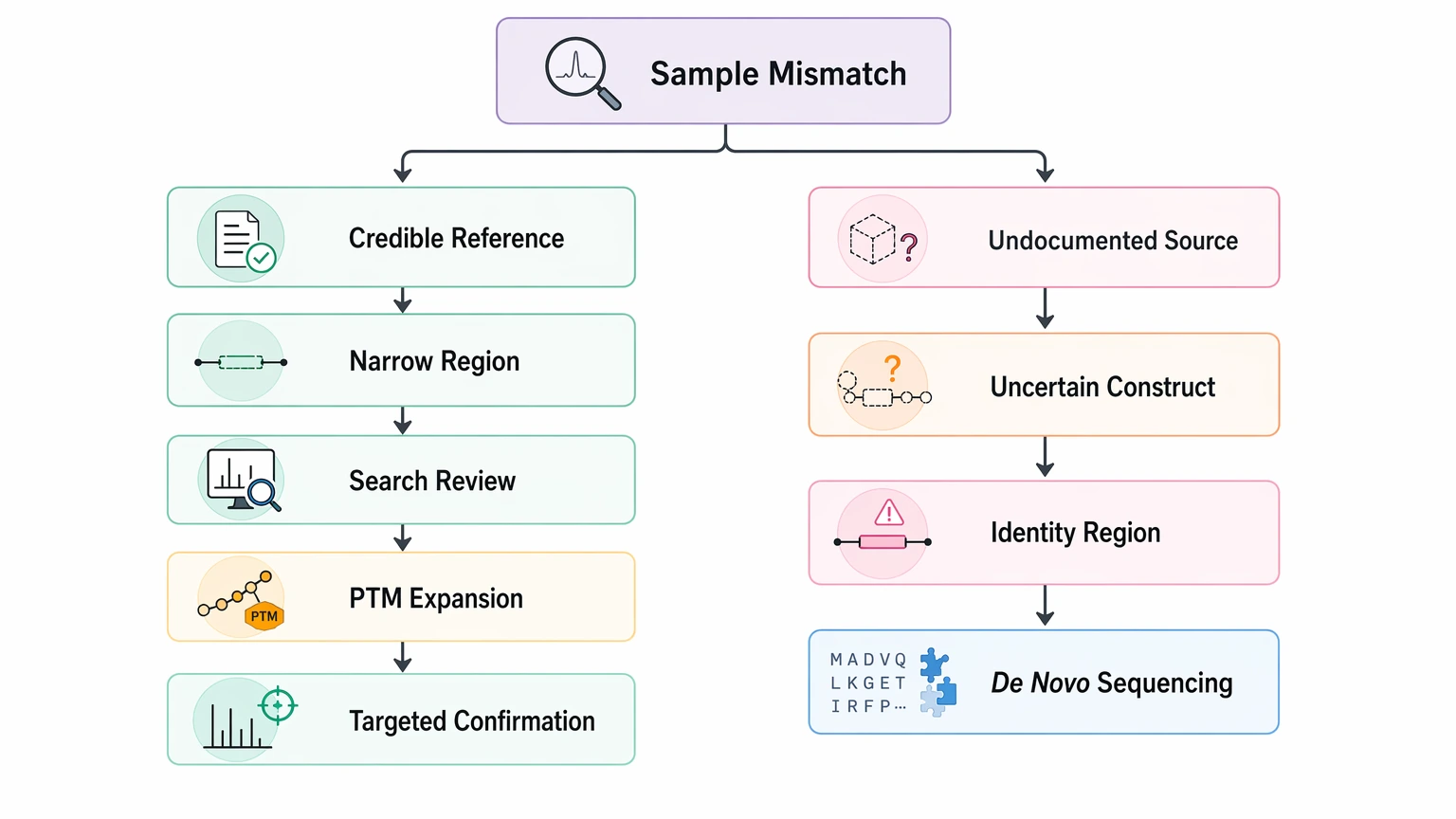
In practical terms, one question usually settles it: Do you need to know what sequence is actually present, or only whether the sample roughly matches an expected entry? The first question supports escalation. The second often does not.
Conclusion
De novo protein sequencing is the right choice when unresolved LC-MS/MS evidence points to a real sequence problem that routine database search cannot explain and the project needs sequence-resolving interpretation rather than another database score. It is especially suitable for undocumented proteins, database-mismatched regions, truncation questions, and sequence variant investigations with interpretable peptide evidence. For protein characterization teams working with uncertain references, mixed historical materials, or biologics anomalies, the most practical next step is to define the decision target, check whether the spectra truly support sequence inference, and then contact us or contact MtoZ Biolabs to evaluate your project in the context of sample quality, likely deliverables, and the right orthogonal validation plan.
FAQ
Can de novo protein sequencing still be useful if only one region is unexplained?
Yes, if that region affects the decision you need to make. A localized mismatch near a fusion boundary, processing site, or suspected truncation may justify region-focused de novo interpretation even when the rest of the protein matches the reference sequence.
Does a low database-search score always mean the protein is novel?
No. Low scores can come from poor fragmentation, incomplete databases, unexpected PTMs, or mixed spectra. Novelty becomes more plausible only after those explanations have been checked and the unmatched evidence still repeats.
How should partial protein sequence reconstruction be reported internally?
Treat it as a bounded result, not a failed one. If the reconstructed region answers the project question, the work may already be enough for follow-up validation or construct review, even without complete sequence coverage.
Are PTM-rich samples good candidates for de novo sequencing?
Sometimes, but not automatically. PTMs can justify escalation because they complicate database matching, yet they can also reduce spectral clarity and make residue-level interpretation harder.
When is de novo peptide sequencing enough without full protein reconstruction?
It is often enough when the goal is to confirm one mismatch region, support a suspected sequence variant, or identify a terminal processing change. Full protein sequence reconstruction is more appropriate when the broader identity of the protein remains uncertain.
What information should be gathered before requesting a sequencing assessment?
Prepare the expected reference sequence if available, peptide mapping outputs, database-search results, intact mass observations, sample source history, digestion details, and the exact decision the project must support.
How to order?

