





What Sample Information Is Needed Before Starting a De Novo Antibody Sequencing Project?
- the sample is labeled only as “antibody” or “IgG”
- concentration is estimated loosely or not recorded
- the purification history is unknown
- the formulation buffer contains unreported additives
- the request asks for “the sequence” without defining whether the goal is CDR confirmation, full antibody variable region recovery, or a re-expression-oriented output
- prior files such as intact mass, SDS-PAGE, or peptide mapping data are available internally but not shared at project start
- CDR identification only
- full heavy chain and light chain antibody variable region recovery
- sequence recovery intended for recombinant re-expression
- comparison against an existing sequence hypothesis
- protein-first sequence recovery with orthogonal support if the purified sample proves ambiguous
- purified monoclonal antibody
- Protein A/G-purified antibody
- antigen-purified antibody
- partially purified antibody fraction
- hybridoma supernatant
- hybridoma cells
- serum-derived antibody fraction
- antibody-containing material with unknown co-purified species
- buffer identity and pH
- salt concentration
- glycerol percentage
- sugars such as sucrose or trehalose
- carrier proteins such as BSA
- detergents or surfactants
- sodium azide or other preservatives
- reducing agents, if present
- co-purified immunoglobulins
- antigen carryover
- degraded fragments
- multiple antibody populations
- low target abundance relative to background proteins
- host species, such as mouse, rabbit, humanized, or chimeric
- known isotype or subclass
- whether the sample is expected to be monoclonal or may contain mixed populations
- known engineered regions
- expected framework region characteristics
- any link to a parental clone or earlier antibody version
- prior LC-MS/MS files
- historical peptide mapping
- intact mass data
- SDS-PAGE images
- Western blot notes
- ELISA specificity notes
- hybridoma availability
- partial NGS reads
- RACE products
- a previous sequence hypothesis from another internal or external effort
- internal plausibility review only
- recombinant expression of candidate heavy chain/light chain pairs
- assay lot transfer
- comparison against a reference clone
- preparation of a re-expression-ready sequence package
- sample type and antibody format
- total amount, concentration, and volume
- sample purity status
- full formulation buffer details, if known
- species, isotype, or engineering background
- project goal: CDR only, full variable-region recovery, or re-expression-oriented output
- any prior LC-MS/MS, peptide mapping, or intact mass data
- availability of hybridoma, NGS, or RACE support
- intended downstream use and validation plan
- any uncertainty labels such as “estimated concentration” or “unknown purification history”
Before you submit a de novo antibody sequencing request, it helps to gather five basics: the starting material type, total sample amount and concentration, sample purity, formulation buffer, and the sequence recovery objective. Those details shape the initial feasibility assessment, affect how cleanly LC-MS/MS can proceed, and reduce ambiguity during chain assignment and heavy/light chain pairing.
In many projects, delays begin with missing context rather than with poor sample quality alone. A purified monoclonal antibody in an unknown buffer, with unclear species background or an undefined deliverable, may still be workable. It just takes longer to review, and the sequencing plan becomes less focused. In practice, a short technical package prepared before submission usually saves more time than sending the tube first and filling in the details afterward.
Where Pre-Submission Planning Usually Breaks Down
This issue often shows up with a legacy antibody reagent that still works in an assay but no longer has a trusted sequence record. The team may need sequence recovery for assay redevelopment, clone transfer, reagent rescue, biosimilar comparison, or recombinant re-expression. The sample exists, but the project package is incomplete.
Common gaps include:
Those gaps affect real planning decisions. They can slow workflow selection, increase uncertainty during heavy chain and light chain interpretation, and lead to mismatched expectations about what the final sequence package is supposed to support.
Why Missing Information Slows Sequence Recovery
A few cause categories matter most here.
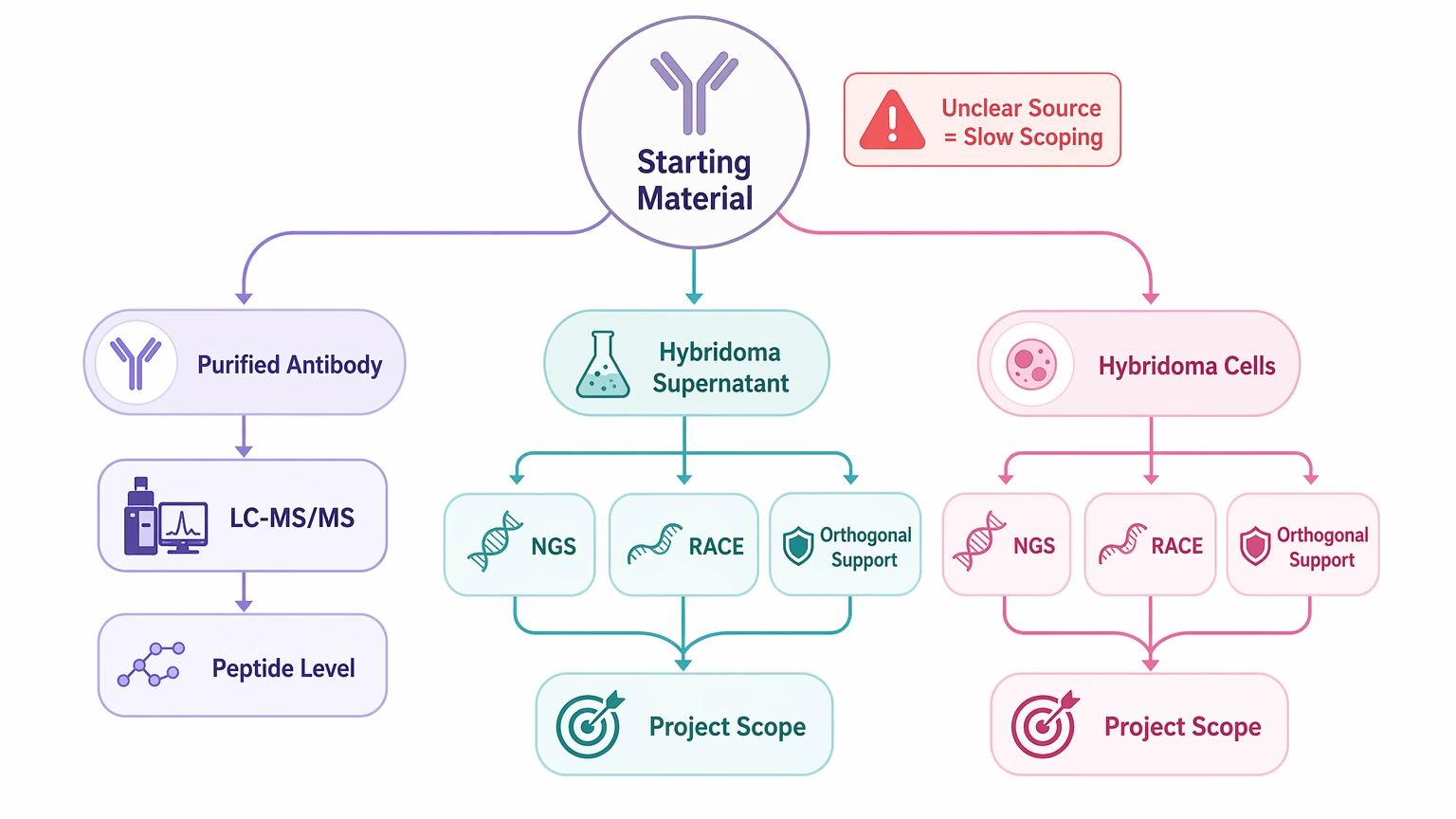
1. The starting material changes the sequencing strategy
A purified antibody, hybridoma supernatant, and hybridoma cells do not support the same workflow. Protein-only input usually relies on mass spectrometry and peptide-level interpretation. Cell-based material may also support NGS or RACE inputs. If the source is not stated clearly, the project cannot be scoped efficiently.
2. Sample condition affects analytical confidence
Low concentration, limited total mass, degradation, and uncertain sample purity do not automatically stop a project. They do change how much confirmatory evidence can be generated and how conservative the interpretation needs to be.
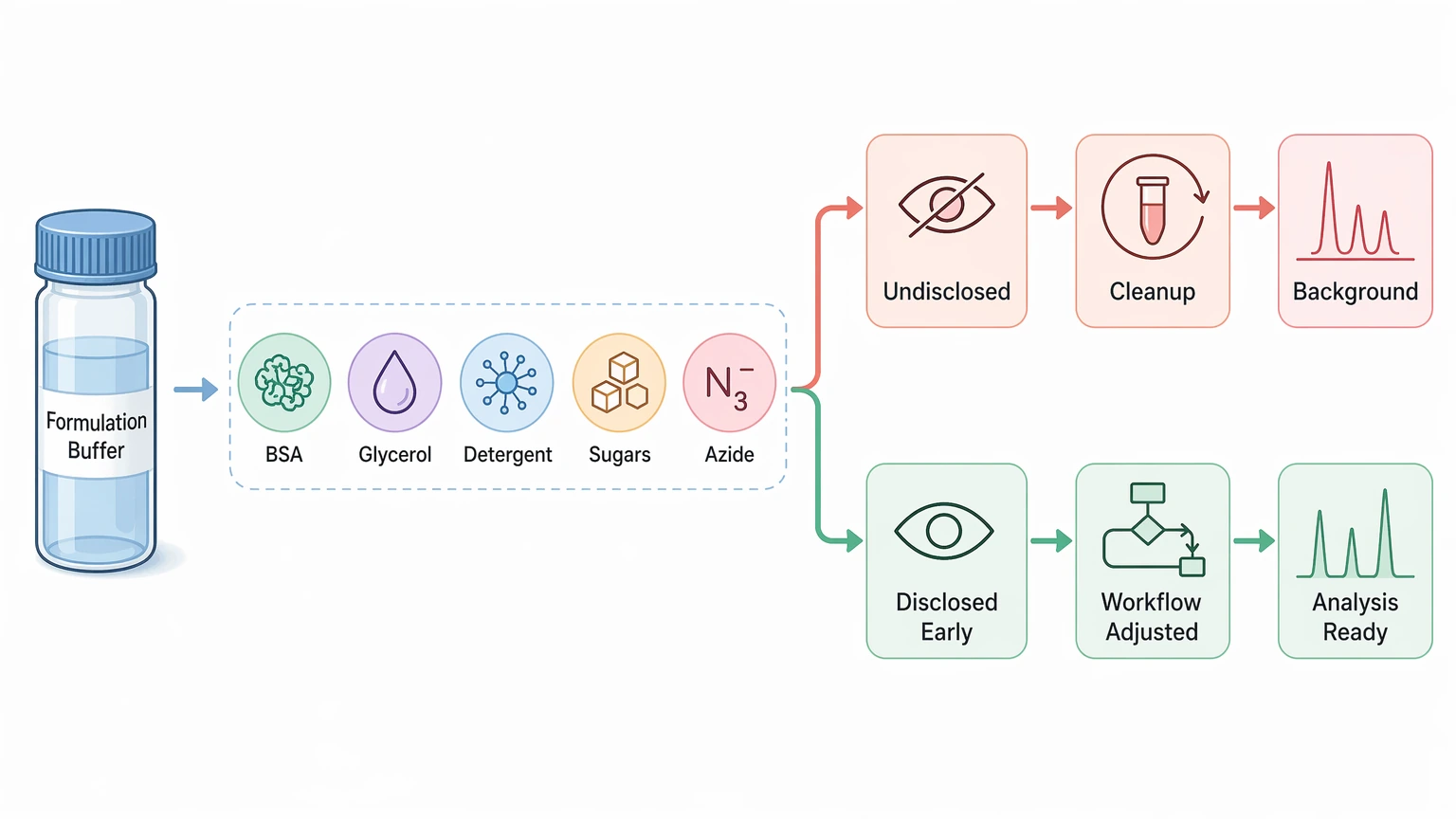
3. Formulation components can interfere with analysis
Additives such as BSA, glycerol, detergents, sugars, azide, and other preservatives can complicate cleanup or introduce background signals. When those components are disclosed early, the workflow can be adjusted before analysis starts.
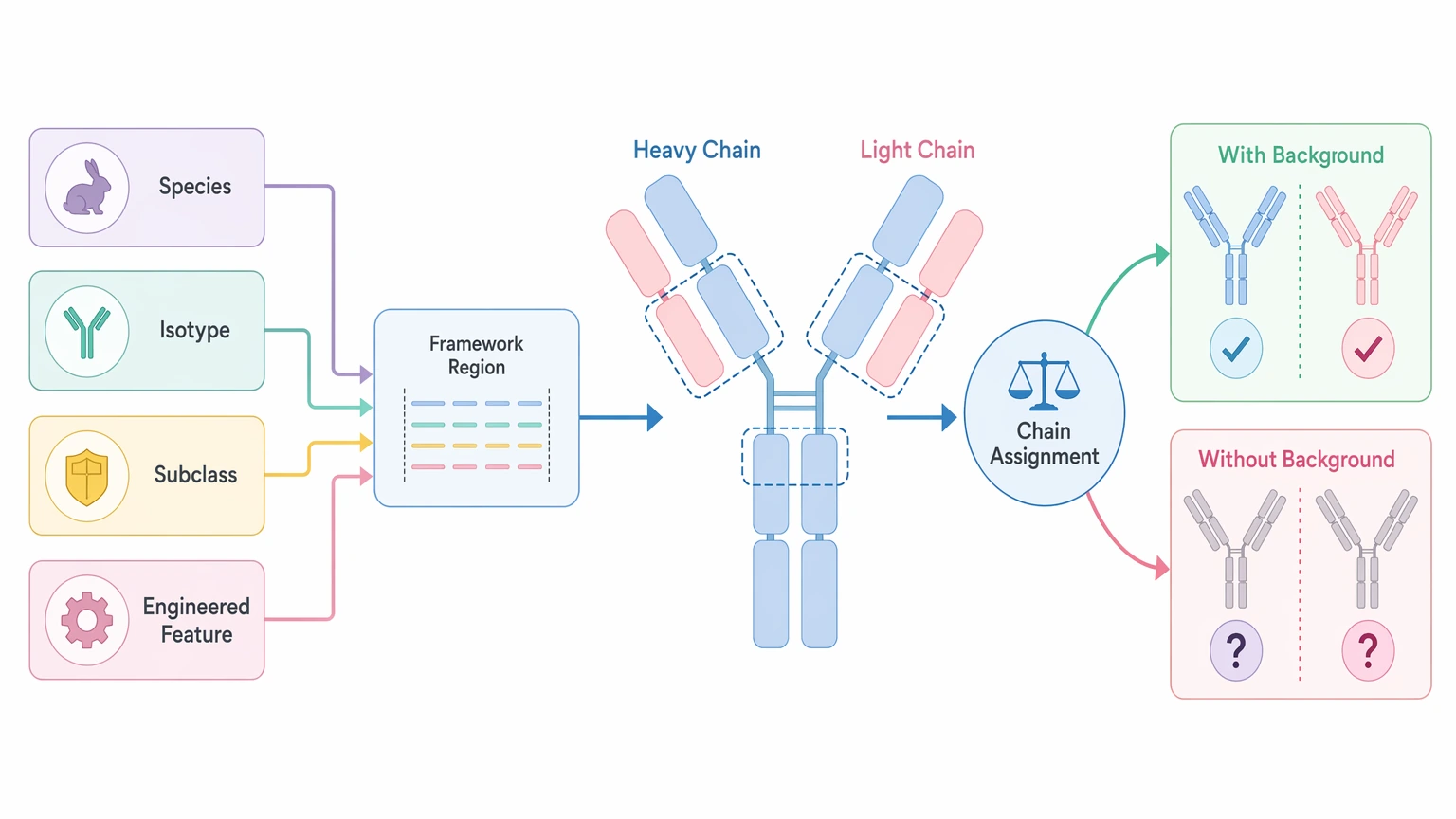
4. Antibody background guides chain assignment
Known species, isotype, subclass, or engineered features help narrow interpretation of the framework region and improve chain assignment logic. Without that background, the analytical data may still be useful, but sequence review becomes less straightforward.
5. A vague deliverable creates avoidable rework
The information needed for CDR identification is not the same as what is needed for full antibody variable region recovery or for downstream recombinant re-expression. If the final use is unclear, the review can start with the wrong scope.
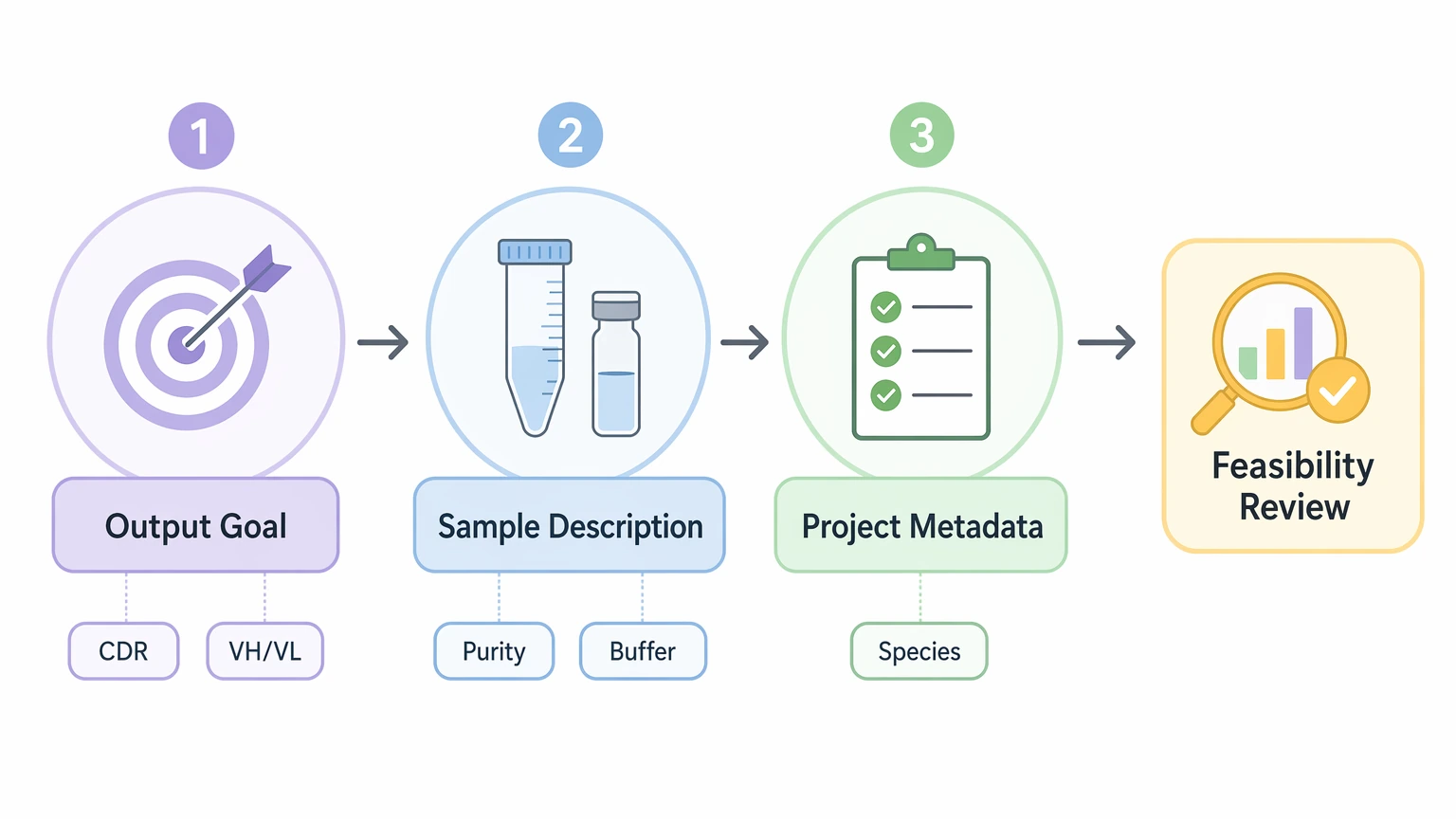
A Project-Planning Guide for Submission Readiness
The most practical sequence is simple: define the output first, describe the sample clearly, then add the metadata that lowers interpretation risk.
Step 1: Define the sequence recovery goal
Start with the decision the recovered sequence needs to support.
Useful goal statements include:
This step matters because the deliverable changes the evidence threshold. A project focused on CDR confirmation can stay narrower than one intended to rebuild the antibody for assay transfer or redevelopment.
A concise objective statement helps, for example:
“Need full VH/VL recovery from a purified monoclonal antibody for recombinant re-expression and assay redevelopment.”
Step 2: Describe the starting material precisely
Next, identify what is actually being submitted.
Useful categories include:
Also note the antibody format when known, such as intact IgG, Fab, scFv-derived reagent, or a more complex engineered construct. Standard architecture usually makes heavy/light chain pairing easier to interpret. Nonstandard formats should be disclosed before analysis rather than discovered during review.
Step 3: Report amount, concentration, and sample purity
Even estimated values are useful when they are labeled honestly.
| Information needed | How to report it | Why it matters |
|---|---|---|
| Total amount available | µg or mg | Helps scope sequence recovery and any repeat analysis |
| Concentration | mg/mL | Supports handling and aliquoting decisions |
| Sample volume | µL or mL | Confirms whether the reported mass is practically usable |
| Sample purity | affinity-purified, partial, unknown | Indicates background complexity |
| Storage condition | frozen, refrigerated, lyophilized | Adds context for stability |
| Freeze-thaw history | none, limited, repeated, unknown | Helps assess degradation risk |
If you already have SDS-PAGE, SEC, or protein quantitation data, include them. They do not replace sequencing evidence, but they improve early judgment about whether the sample state matches the stated project goal.
Step 4: Disclose the formulation buffer and likely contaminants
This is one of the best ways to avoid delays.
List the formulation buffer as completely as possible, including:
Also flag contamination risks that may complicate interpretation:
If your sample includes formulation complexity or limited material, this is the right point to submit your requirements for review. In that setting, MtoZ Biolabs can evaluate whether the available sample package is better suited to a mass-spectrometry-first workflow alone or whether extra support material should be considered before intake.
Step 5: Add biological background that supports chain assignment
Not every legacy reagent comes with complete records, but a few details can make interpretation easier.
The most useful items are:
These details do not override analytical evidence. They help reviewers assess whether proposed peptide-to-chain mapping is biologically plausible.
Step 6: Share orthogonal data and supporting materials
Teams sometimes hold back partial data because it seems incomplete. In practice, even incomplete support can improve planning.
Useful supporting inputs include:
These materials may support orthogonal validation or show that purified protein alone is not the strongest starting point.
Step 7: State what happens after sequence recovery
The downstream use should be explicit before the project starts.
Examples include:
That context changes how sequence interpretation is reviewed. If the project is moving toward redevelopment rather than basic identification, say that upfront. A decision-stage conversation is also a practical place to contact us with your sample facts and intended use case; MtoZ Biolabs can then evaluate the project around sequence recovery, chain assignment risk, and follow-up validation planning.
When Purified Antibody Alone May Be Insufficient
A purified sample can be a valid starting point, but several situations increase ambiguity:
| Scenario | Why it is harder | Helpful next input |
|---|---|---|
| Very low sample mass | Limits repeat analysis and confirmation options | More material or prior LC-MS/MS data |
| Mixed antibody population | Complicates peptide-to-chain interpretation | Hybridoma source or clone-separated material |
| Heavy formulation background | Increases cleanup burden and background interference | Full buffer disclosure or a reformulated aliquot |
| Nonstandard architecture | Makes pairing logic less direct | Construct notes or parental design context |
| Re-expression-ready output required | Raises the bar for interpretable sequence recovery | Hybridoma, NGS, or RACE support |
These cases do not mean the project should stop. They mean the feasibility discussion should include contingency paths instead of assuming one input type will answer every sequence question.
A Practical Submission Checklist
Before sending the sample, confirm that your package includes:
Honest uncertainty helps. A clearly marked unknown is easier to plan around than a blank field or an unsupported assumption.
Conclusion
For de novo antibody sequencing, readiness is defined less by any single sample metric than by the combination of sample identity, condition, formulation, antibody background, and intended use. That framework is especially useful for legacy monoclonal antibody recovery, assay redevelopment, clone transfer, and early-stage recombinant re-expression programs where chain assignment clarity matters as much as raw sequence output. If your team can provide a purified sample together with the core metadata listed above, the project usually enters review with a clearer path to sequence recovery. If key details are still missing, a pre-submission consultation can help determine what should be clarified or added before sequencing begins.
FAQ
Do I need to know the antibody target before starting de novo antibody sequencing?
No. Target identity can help frame downstream use, but it is not required for every project. More useful starting information is the sample source, purity, formulation buffer, and whether you need CDR-focused or re-expression-oriented sequence recovery.
Is a concentration estimate acceptable if I do not have a recent quantitation result?
Yes, as long as you label it as an estimate and include how it was derived. An approximate mg/mL value together with sample volume is usually more useful than leaving the field blank.
Should reduced and non-reduced SDS-PAGE both be shared if available?
Yes. Paired gel views can add context about chain integrity, fragmentation, and sample complexity before LC-MS/MS work is planned.
If I suspect the sample contains more than one antibody, should I still submit it?
You can, but state that clearly. A suspected mixed population changes the feasibility discussion, especially if your goal is full heavy chain/light chain sequence recovery rather than a limited CDR readout.
What if I have hybridoma cells but want to start from purified antibody first?
Mention both. A protein-first workflow may still make sense, but knowing that hybridoma material exists gives the project a stronger fallback path if purified antibody data alone leaves unresolved pairing or framework ambiguities.
Does re-expression planning require more than a sequence summary?
Usually, yes. If the final goal is recombinant redevelopment, the project should be framed around interpretable heavy chain/light chain sequence recovery, plausibility review, and an orthogonal validation plan rather than a minimal sequence note.
How to order?

