





Sample and Input Requirements for Monoclonal Antibody Sequencing Projects
- Minimum useful input: purified monoclonal antibody with known concentration and buffer details
- Better input set: purified antibody plus species, isotype/subclass, purification history, and prior analytical records
- Best fit for de novo work: material that supports chain-aware digestion, PTM review, and follow-up confirmation where needed
- Important limitation: standard MS/MS interpretation may not fully resolve every CDR residue, glycoform-affected peptide, or leucine/isoleucine position from a single submission
- Is the antibody enriched relative to other proteins?
- Is the concentration known well enough for digestion planning?
- Has the material been freeze-thawed repeatedly?
- Does the formulation contain salts, sugars, glycerol, surfactants, preservatives, amino acids, or carrier proteins such as BSA?
- Is the sample intact, reduced, or available in both states?
- species
- isotype or subclass
- expression system, if known
- clone identifier and transfer history
- purification method
- formulation buffer
- prior SDS-PAGE, SEC, or intact mass observations
- previous peptide mapping or LC-MS/MS results
- suspected post-translational modification (PTM) patterns
- intended use of the final sequence
- reconstructed heavy chain and light chain sequence proposals
- annotated sequence coverage
- local confidence or confidence score reporting
- PTM and glycoform notes where supported
- a list of unresolved or low-confidence positions
- comments on database-search limitation and evidence gaps
A team preparing for monoclonal antibody sequencing usually needs more than a single tube of purified IgG. The strongest starting package combines an analytically usable antibody sample, key antibody background such as species and isotype, formulation and purification notes, and a clearly defined sequencing objective. That combination creates a more practical path to de novo peptide sequencing and de novo protein sequencing by LC-MS/MS, especially when no confirmed reference sequence is available.
Protein-only submissions can still work, but the risk of local uncertainty is higher in the regions that often matter most: complementarity-determining region (CDR) peptides, glycosylated Fc peptides, disulfide-linked regions, and sites affected by leucine/isoleucine ambiguity or other isobaric residue ambiguity. A useful request should therefore spell out what level of sequence coverage, local confidence, and unresolved positions would still support the downstream decision.
Quick project decision block
Why These Projects Often Stall Before They Start
Many sequencing requests begin after construct records are lost, clone transfer is incomplete, or legacy material has to be reused for functional studies, patent review, or biosimilar comparison. In that situation, teams often expect the antibody protein alone to provide a complete answer. The real challenge is that antibodies are multi-chain, modified proteins, so sequence reconstruction is more involved than a generic single-protein analysis.
The hardest parts are usually not the conserved constant region segments. They are the variable region peptides, especially CDR-containing peptides, along with peptides affected by glycosylation, clipping, oxidation, deamidation, or incomplete fragmentation. A database-search limitation becomes clear when the closest reference does not capture clone-specific differences or when no suitable database entry exists. That is why pre-project planning matters: it reduces avoidable ambiguity before data review starts.
What Counts as a Usable Starting Sample
For most monoclonal antibody sequencing projects, the core input is purified intact antibody protein. “Purified,” though, should mean more than a labeled tube. The sample should provide enough material for digestion, replicate preparation if needed, and at least limited follow-up confirmation. Exact quantity thresholds vary by workflow, but low-volume or low-concentration submissions leave less room for repeat analysis if difficult regions appear.
Sample condition also affects interpretation. The main questions are:
These details influence digestion performance, precursor selection, and the quality of fragment ion evidence. A buffer that works for storage is not always ideal for LC-MS/MS. In the same way, a sample that looks clean by a basic protein assay may still contain excipients that complicate peptide detection or deconvolution.
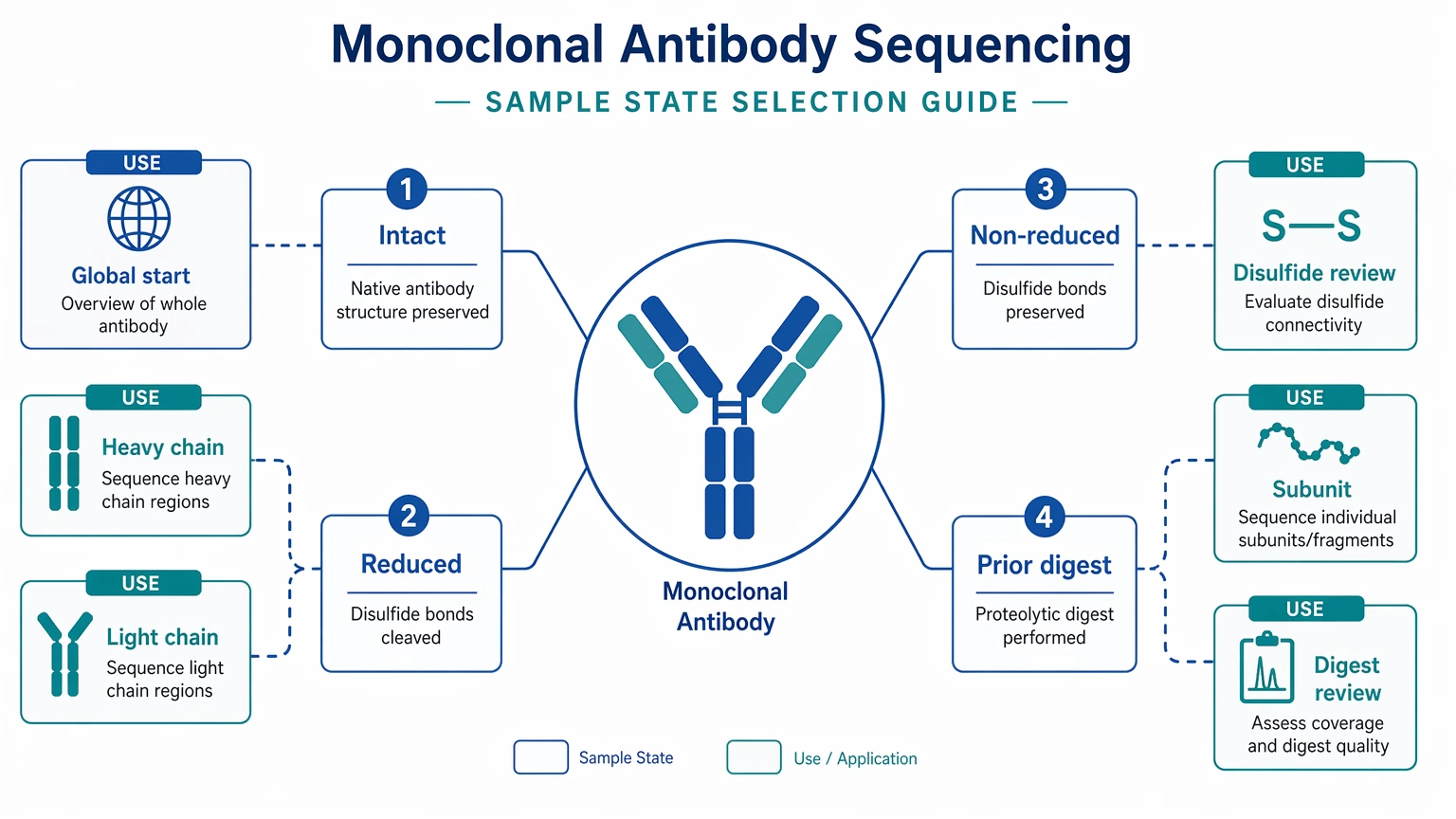
When material is available in more than one form, say so early. Intact antibody is a common starting point. Reduced material may simplify heavy chain and light chain interpretation after reduction and alkylation. Non-reduced material can support disulfide bond-aware review or subunit analysis. If only a prior digest remains, that can still be useful, but the project team should know how it was prepared and whether re-optimization is possible.
Which Background Records Improve De Novo Interpretation
Non-sample inputs often make the difference between a broad search and a focused sequencing plan. Useful records include:
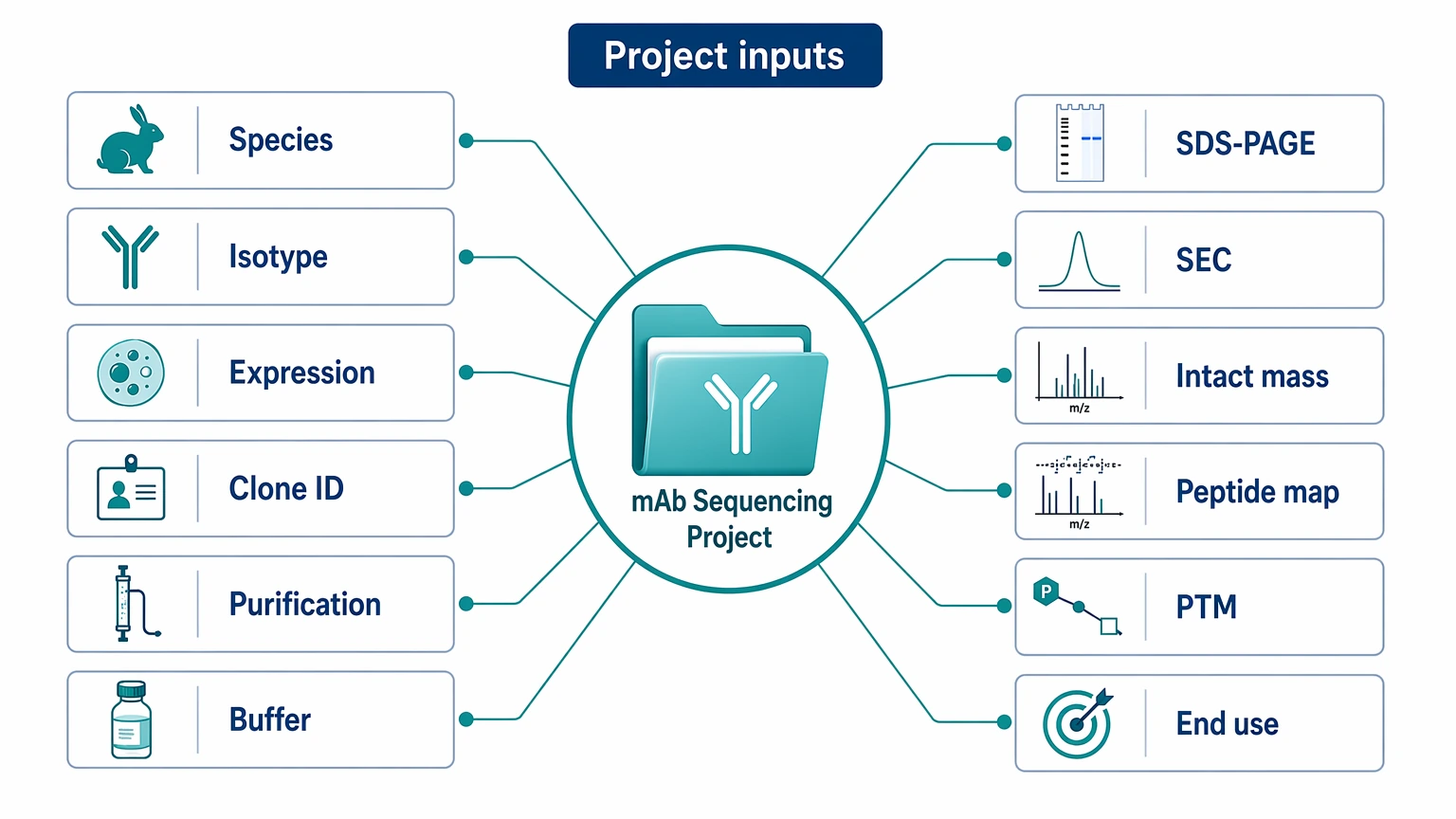
This information narrows the expected antibody architecture and helps separate framework-supported assignments from novel sequence features. For example, known subclass information can guide interpretation of the constant region, while prior intact-mass data may show whether clipping or unexpected mass shifts are already present. Formulation notes can also explain signal suppression or digestion issues before those effects are mistaken for sequence gaps.
A useful planning discussion should also define the real project objective. Some teams need full chain reconstruction. Others mainly need variable-domain recovery, similarity assessment between two antibodies, or a confidence-ranked consensus sequence that is sufficient to guide re-engineering. When the success criterion is vague, the report can end up answering the wrong question.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
Step-by-Step Project Planning for Antibody Sequencing
The most effective planning sequence is not a generic “send sample and wait” workflow. For antibody projects, the steps should reflect the structure of the molecule and the main interpretation risks.
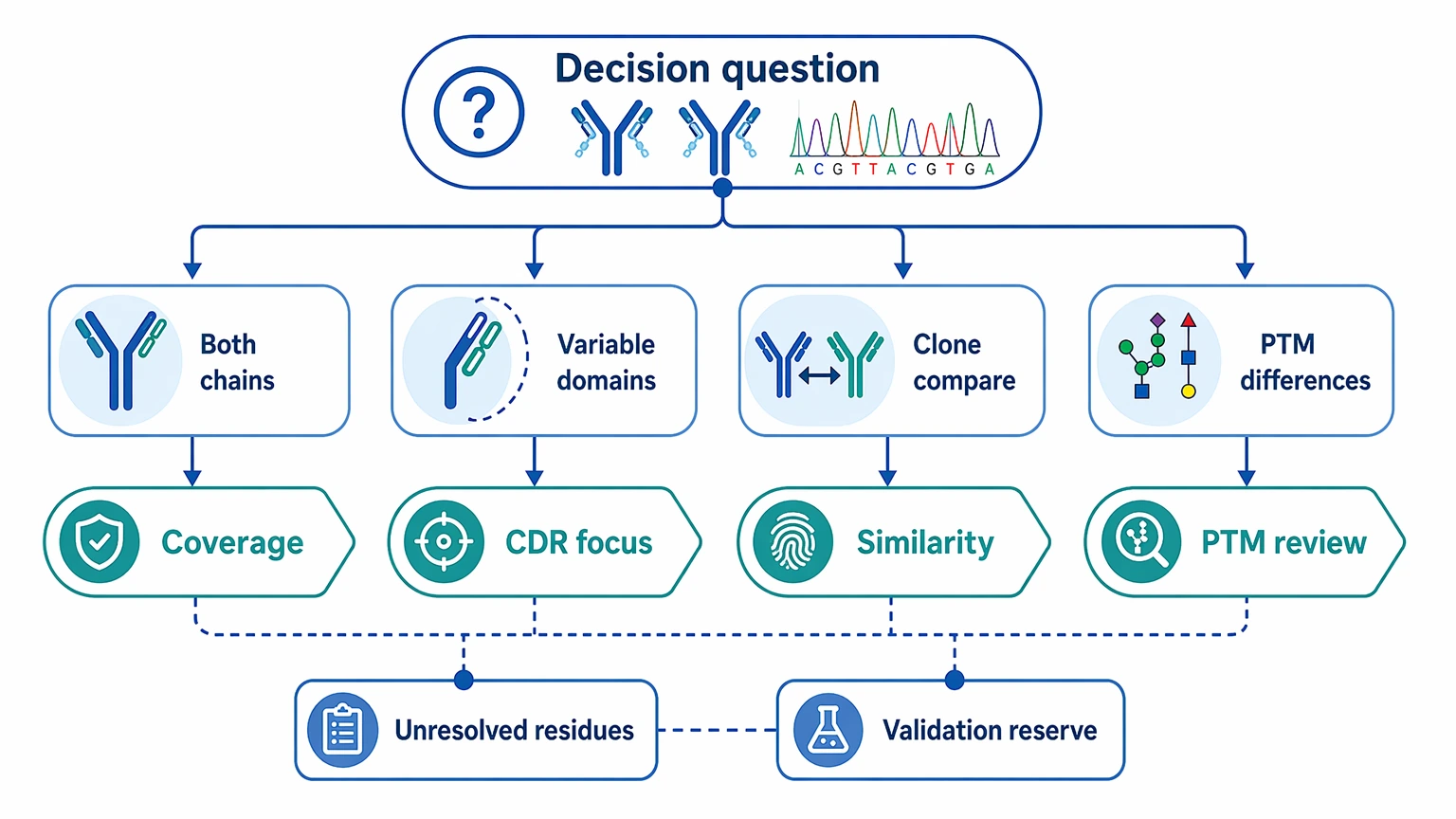
Step 1: Define the decision question.
State whether the project is meant to reconstruct both chains, recover only the variable domains, compare related clones, or document PTM-aware differences. That decision sets the tolerance for unresolved residue calls and determines whether follow-up validation should already be reserved.
Step 2: Match the sample state to the goal.
If chain reconstruction is the goal, intact material plus reduced material is often more informative than a single presentation state. If disulfide context or clipping is a concern, include non-reduced material or request intact mass / subunit analysis up front.
Step 3: Disclose anything that can interfere with peptide interpretation.
Surfactants, salts, sugars, serum carryover, carrier proteins, or mixed lots should be flagged before launch. These factors can reduce peptide readability and increase the chance that low-confidence calls cluster in the wrong places.
Step 4: Clarify whether the workflow is truly de novo or partly reference-informed.
A project can remain fundamentally de novo while still using species, isotype, or framework context to narrow the search space. That support is often useful, but it does not remove ambiguity in novel CDR regions.
Step 5: Ask for reporting that reflects uncertainty honestly.
The requested deliverable should include chain-level assignments, local confidence, notes on leucine/isoleucine ambiguity, PTM annotations, and any gap region or provisional sequence segment. If the project may require extra evidence, ask the team to outline a validation strategy before sample consumption becomes limiting.
When the available material is incomplete or the target outcome is narrow, a scoped planning review is often the best next step. Teams that need to align sample condition, chain-specific workflow options, and reporting criteria can submit your requirements to MtoZ Biolabs for project evaluation before committing limited antibody material.
Expected Results and Validation Methods
A well-prepared project should improve interpretability, not imply universal full-sequence resolution. The first deliverables are usually the direct outputs of the primary sequencing workflow:
Those are the immediate deliverables. Follow-up confirmation is different and should be treated separately. It may include targeted LC-MS/MS review of low-confidence peptides, orthogonal peptide mapping, repeat digestion, additional subunit analysis, or comparison against another lot or related sample. In other words, the first report should show what is well supported now and what still needs confirmation.
Validation should be prioritized when the uncertain residues sit in a variable region, change a CDR interpretation, or affect clone discrimination. If the remaining uncertainty is confined to non-critical positions, a documented consensus sequence may still be suitable for the intended research use.
Key Cautions and Practical Limits
A few constraints should be discussed before submission.
Sample quality or amount limits: very limited material leaves less room for repeat digestion, replicate injections, or targeted follow-up. If sample is scarce, rank the primary question first.
Controls and repeat expectations: a previous lot, related antibody, old peptide map, or intact-mass result can help distinguish true sequence differences from incomplete evidence. Repeat analysis may still be needed when critical residues remain ambiguous.
Batch or contamination risk: mixed lots, serum proteins, BSA, excipients, or surfactants can distort peptide-level interpretation. Cleanup steps may help, but they can also consume material.
Interpretation boundaries: standard MS cannot reliably separate every leucine from isoleucine, and heavily modified peptides may produce incomplete fragmentation. Some PTM-rich or glycosylated regions may therefore remain provisional after the first pass.
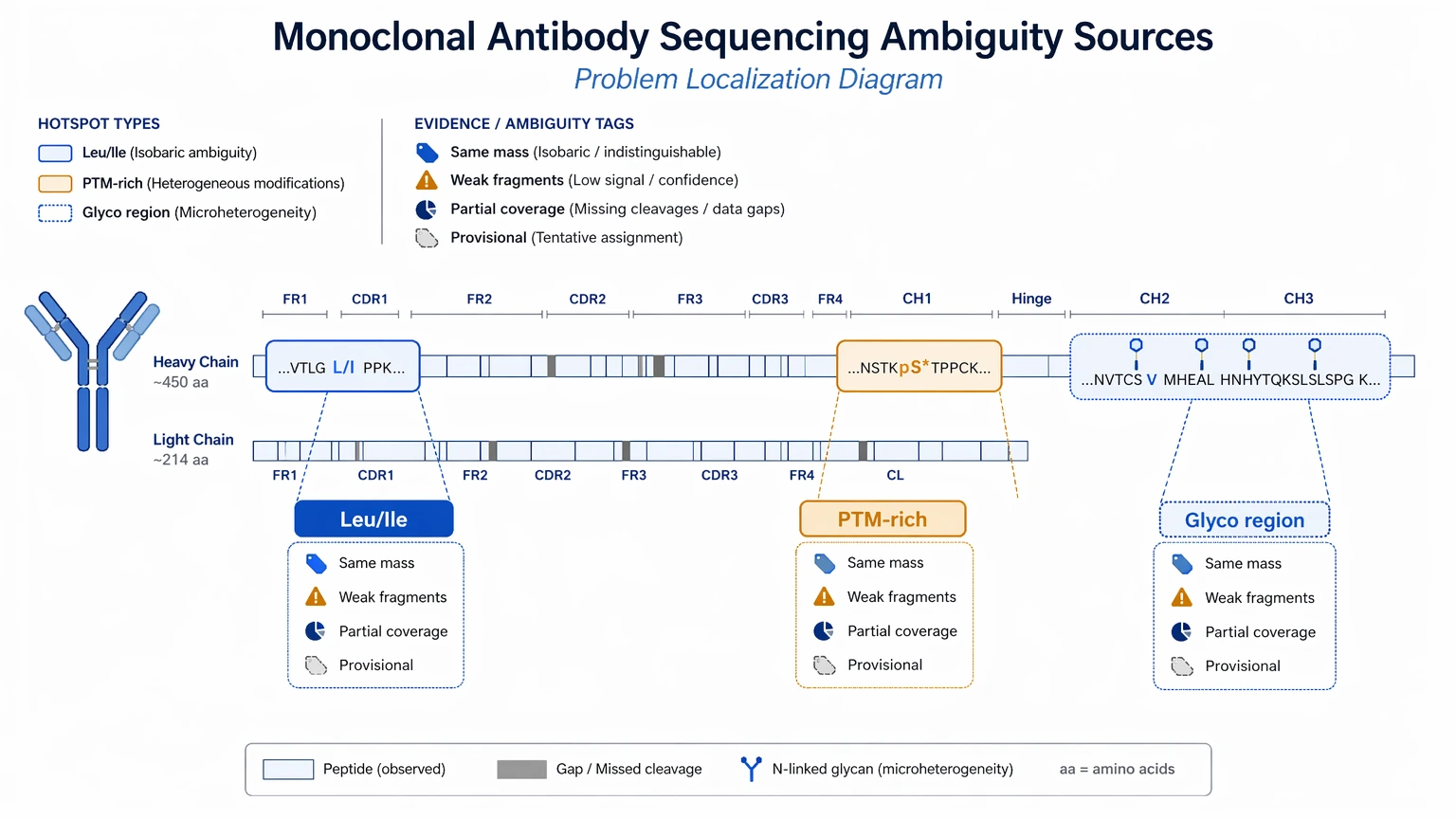
When another method or outside support is better: if the project requires absolute residue identity in a narrow region, direct confirmation using targeted validation or orthogonal molecular information may be the better next step than extending de novo interpretation alone.
Conclusion
A realistic request starts with three things: usable antibody material, records that narrow the antibody context, and a defined success criterion for chain reconstruction or variable-region interpretation. That framework is especially helpful for legacy clones, missing construct histories, and antibody comparison studies where de novo protein sequencing has to work around formulation effects, PTM burden, and local ambiguity. If your team needs to decide whether the current sample package is sufficient, contact us at MtoZ Biolabs to evaluate your project in the context of antibody sequencing workflow choice, expected reporting, and practical follow-up confirmation.
FAQ
If I only know the clone name, should I still start the project?
Yes, but the clone name alone rarely provides enough context for efficient planning. Even partial records such as host species, purification method, or an older QC sheet can reduce interpretation uncertainty.
Is Fc glycosylation mainly a PTM issue, or does it affect sequence reconstruction too?
It affects both. Fc glycosylation changes peptide behavior during digestion and MS/MS analysis, so it can interfere with residue-level interpretation around glycosylated motifs as well as PTM annotation.
When is a consensus sequence good enough for downstream work?
A consensus sequence is often usable when the research goal is framework comparison, clone grouping, or early re-engineering review and the unresolved positions do not sit in decision-critical CDR sites.
Should I provide prior intact mass data even if it looks low resolution?
Usually yes. Even limited intact-mass information can reveal clipping, heterogeneity, or major mass shifts that shape the sequencing plan and help explain later peptide-level findings.
What is the biggest mistake in defining the project scope?
The most common mistake is asking for a “full sequence” without stating whether the real need is chain reconstruction, clone comparison, or confirmation of a few variable-region residues.
When should I pause and request pre-project review instead of shipping immediately?
Pause when the sample contains stabilizers or carrier proteins, when chain state is unclear, when only a very small amount remains, or when one uncertain CDR position could change the downstream decision.
How to order?

