





Ngs vs Sanger Sequencing: Method Selection and Research Use Cases
- clone verification after plasmid construction
- amplicon confirmation for a known locus
- confirmation of a suspected mutation in a short region
- checking whether a construct carries the intended coding change
- validation of a small number of samples without multiplexing needs
- sequencing many targets or samples in parallel
- detecting low-frequency variants in a mixed population
- comparing multiple strains, constructs, or conditions
- expanding transcript inference in RNA-based workflows
- capturing broader variation than a single targeted assay can provide
-
Processing and modification of the mature analyte. Alternative splicing, RNA editing, signal peptide cleavage, and PTMs can separate the mature peptide or protein from the predicted translation.
-
Heterogeneous sample composition. A purified band or fraction may still contain multiple related forms, including an isoform, truncation, or partially processed species.
-
Weak annotation or novelty risk. In a non-model organism, NGS may generate sequence candidates while downstream assignment remains uncertain because the relevant protein is poorly annotated or represents a novel sequence.
-
Proteomics evidence that does not map cleanly. If database search results show few confident peptide-spectrum match assignments, substantial unmatched spectra, or peptide masses that conflict with the predicted product, additional DNA confirmation may not resolve the main issue.
Quick Answer
Sanger sequencing is the practical first choice for focused confirmation of a known DNA region, such as clone verification, amplicon confirmation, or checking a suspected coding variant in a clean template. NGS is the better fit when the project needs broader discovery, greater sequencing depth, more coverage, or analysis of a mixed population. If the real decision depends on which peptide or protein form is present in the sample, neither DNA method is usually sufficient on its own. In that setting, LC-MS/MS with de novo peptide sequencing or de novo protein sequencing is often the more direct next step.
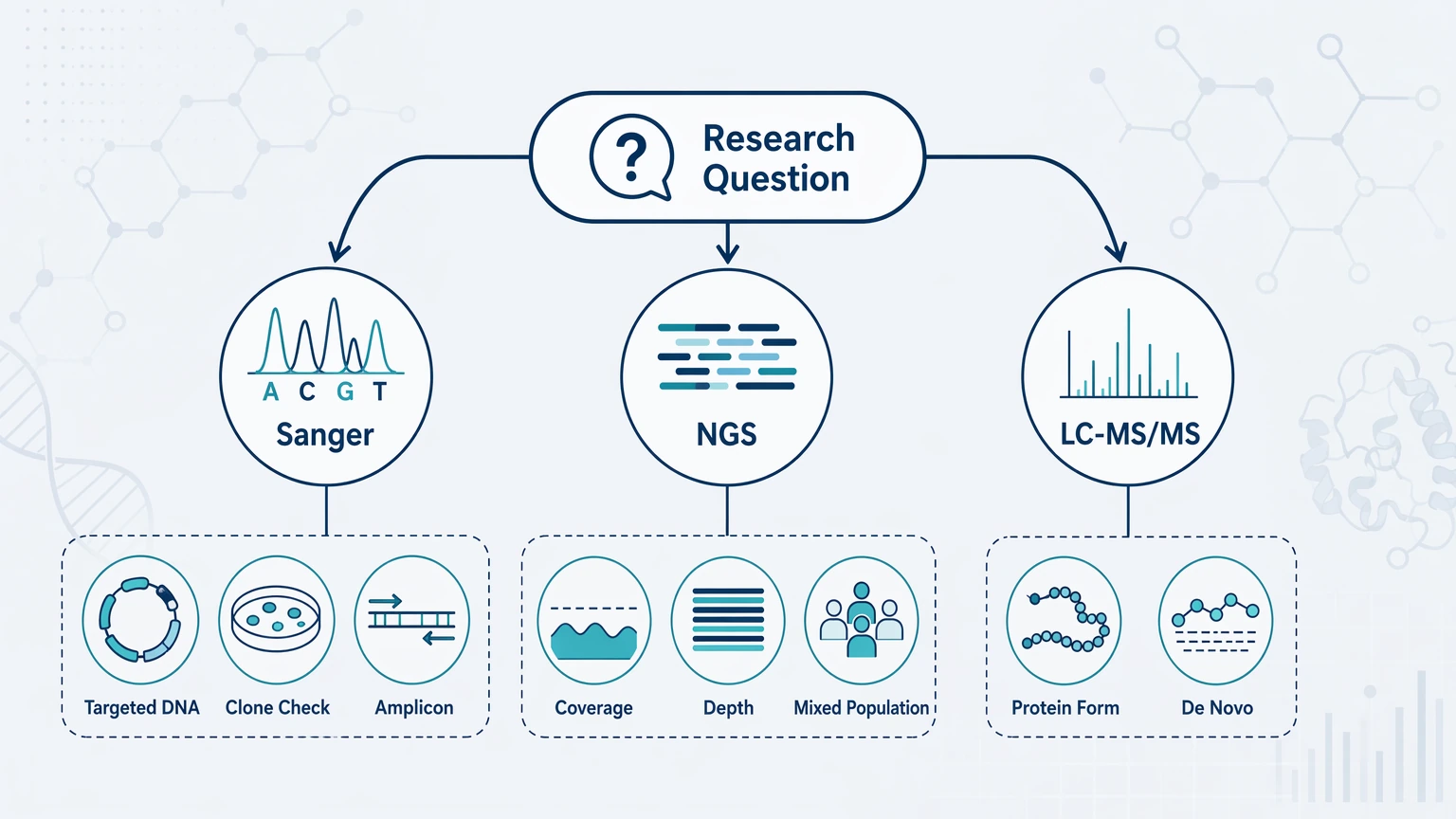
For a quick project screen:
Question is about a DNA template? Start with Sanger sequencing for a small targeted region or NGS for broader or mixed-template projects.
Question is about the molecule measured in the sample? Move toward LC-MS/MS and consider de novo peptide sequencing or de novo protein sequencing.
Transcript or coding sequence exists, but protein data do not fit? A combined workflow is usually more informative than more DNA confirmation alone.
What NGS and Sanger Sequencing Actually Confirm
The first distinction is simple: Sanger sequencing and NGS read nucleic acids, not mature proteins. They can confirm a coding sequence, support transcript inference, and help define an open reading frame. They do not directly show that the protein in the tube matches the predicted translation.
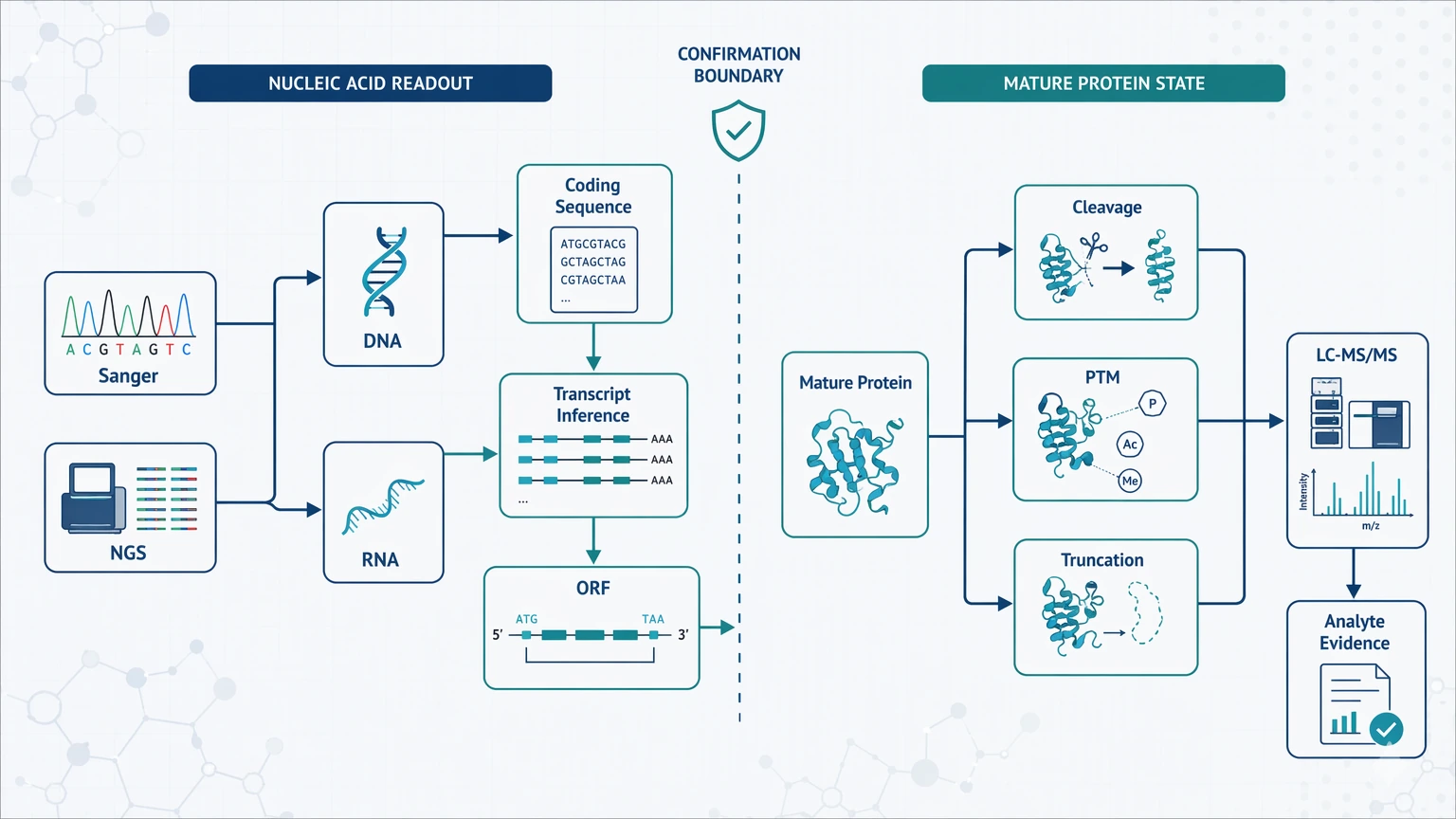
That boundary becomes important as soon as a study moves beyond template confirmation. A construct may contain the expected insert while the expressed product still shows signal peptide cleavage, truncation, PTMs, or multiple processed forms. NGS can also identify candidate transcripts, but protein inference becomes less secure when splicing, RNA editing, weak annotation, or sample heterogeneity changes the final analyte.
For method selection, the more useful opening question is not which platform is stronger in general. It is whether the task is to verify a nucleic acid template or to identify the peptide or protein that was actually measured.
When Sanger Sequencing Is the Better Choice
Sanger sequencing works best for narrow confirmation tasks with a defined expected answer and a mostly single-template sample.
Its main advantage is interpretability. When the template is clean, the chromatogram can often answer a limited question directly without the overhead of a broader sequencing workflow. The tradeoff is equally clear: Sanger sequencing is not suited to complex mixtures, low-frequency variants in a mixed population, or discovery across many loci. It also cannot show whether the final expressed analyte includes processing events or PTMs that change the mature sequence.
When NGS Is the Better Choice
NGS is usually the better choice when the project calls for scale, heterogeneity analysis, or discovery rather than simple confirmation.
Because NGS produces both depth and breadth, it can reveal subpopulations or sequence diversity that a single targeted readout may miss. Even so, it remains a nucleic-acid method. Defining a likely coding sequence or transcript set still does not prove which mature protein form is present after cleavage, processing, or modification.
Where Both DNA Methods Stop Answering the Protein Question
The comparison changes once the decision centers on the analyte rather than the template. Four recurring cause categories usually mark that transition.
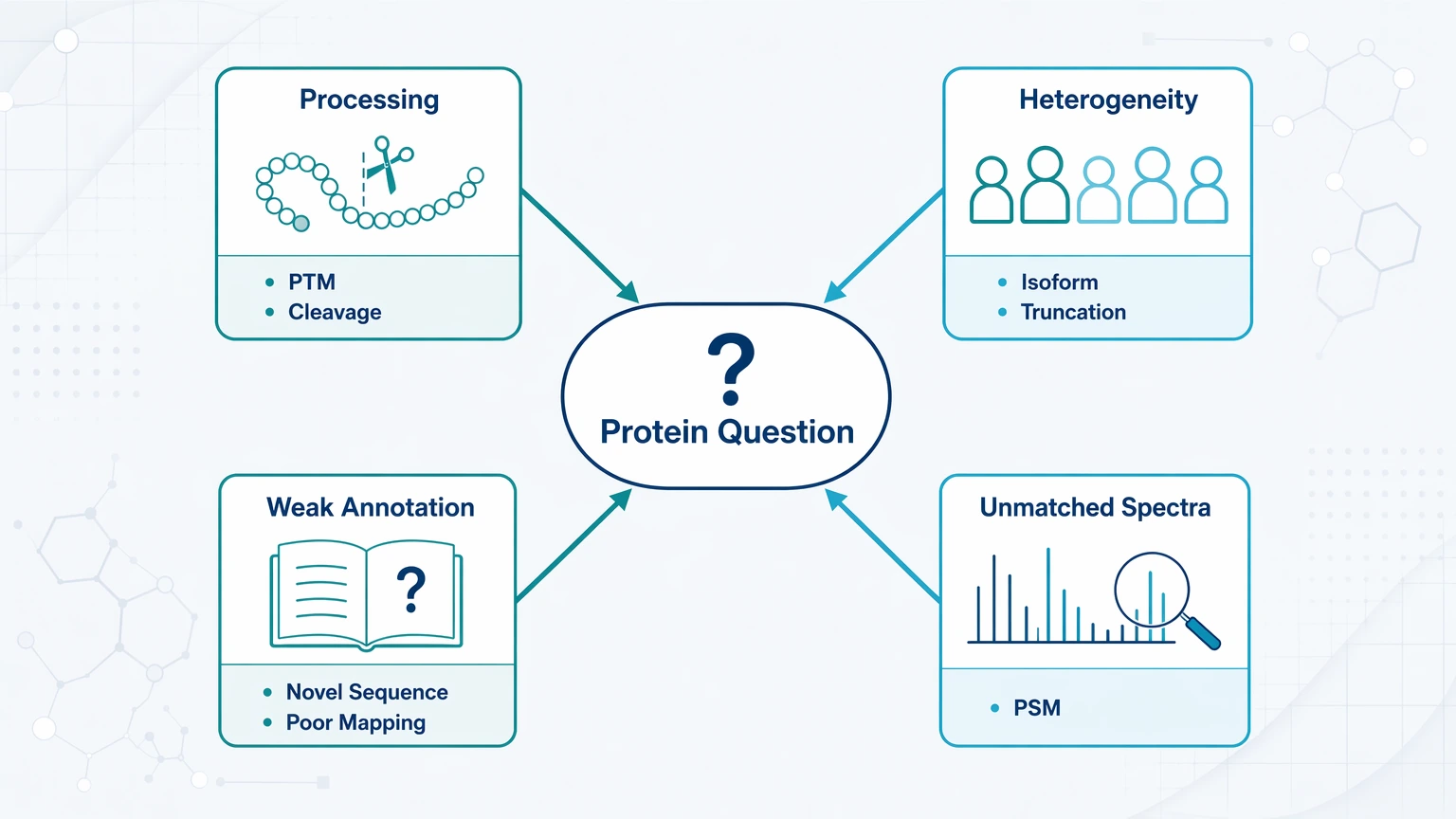
This is the practical boundary: Sanger sequencing and NGS answer template questions well, but they do not directly establish every protein form present in a sample.
How to Decide Whether to Escalate to LC-MS/MS-Based De Novo Sequencing
A strong reason to escalate is disagreement between nucleic-acid expectations and protein evidence. When the observed intact mass does not fit the predicted translation, or when LC-MS/MS data include persistent unmatched spectra, an analyte-level strategy becomes more relevant.
The table below is a simple workflow view for first-pass method selection.
| Scenario | Recommended workflow | What it supports | Key limitation |
|---|---|---|---|
| Edited plasmid or short locus needs confirmation | Sanger sequencing | Efficient targeted sequencing for a defined region | Does not show the mature protein form |
| Many loci, many samples, or heterogeneous templates | NGS | Scale, depth, and mixed-population analysis | Protein identity remains inferred |
| Candidate transcript found, but protein masses disagree | NGS plus LC-MS/MS | Links transcript inference with analyte evidence | May still require de novo interpretation |
| Unknown purified protein with weak database search results | De novo protein sequencing | MS/MS fragmentation and sequence tag evidence from the analyte | Some residues may remain ambiguous |
| Novel peptide with no strong database hit | De novo peptide sequencing | Direct analysis for short analytes absent from reference databases | Sequence continuity may be incomplete |
The main takeaway is that escalation is justified when the unresolved question has moved from template identity to analyte identity. In that transition zone, readers can submit your requirements to MtoZ Biolabs for workflow evaluation when the issue involves unmatched spectra, intact-mass mismatch, PTM burden, or uncertainty about whether DNA sequencing is still the right next step.
Expected Results and Validation Methods
Different methods produce different forms of evidence, so the expected deliverable should match the research goal.
For Sanger sequencing, the immediate output is targeted sequence confirmation for the selected region, usually with chromatogram-based interpretability. For NGS, the output is broader sequence evidence, coverage, sequencing depth, and support for variants, subpopulations, or transcript candidates. For de novo peptide sequencing or de novo protein sequencing, the output is analyte-level interpretation from LC-MS/MS, such as sequence tags, candidate residue assignments, PTM observations, unmatched regions, and a stated level of sequence confidence.
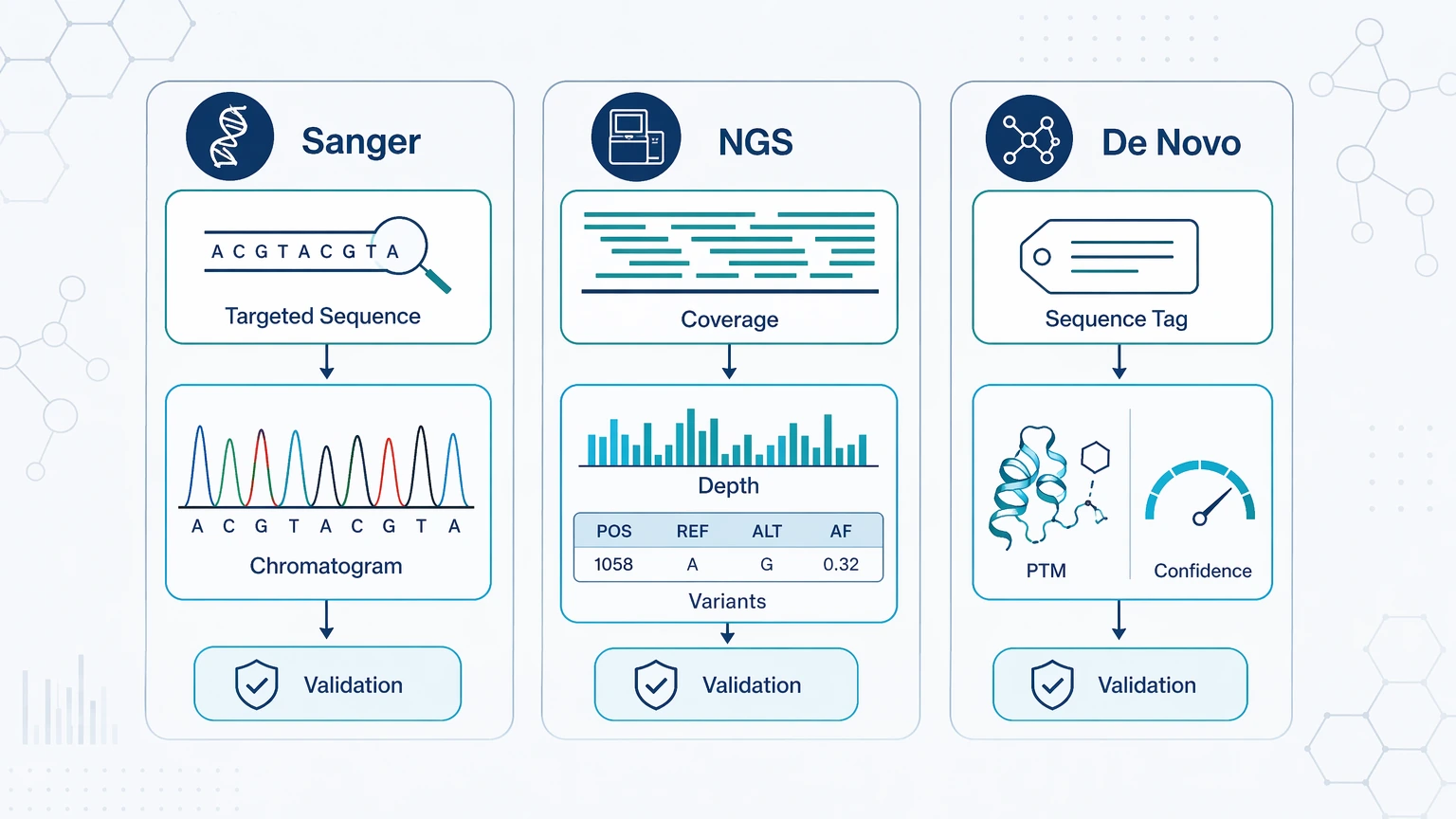
Validation remains method-specific. Follow-up may include repeat amplification, a second primer set, targeted resequencing, targeted MS validation, terminal sequencing, intact-mass comparison, or another orthogonal validation step. One limit should stay explicit: MS/MS interpretation does not always produce a single unambiguous full sequence. PTMs, incomplete fragmentation, database-search limits, and isobaric residue ambiguity can leave some positions unresolved.
Service Routes to Consider
If your project has moved from template confirmation to analyte resolution, these routes are often the most relevant next checks.
Key Cautions and Practical Limits
Method selection becomes clearer when the practical constraints are stated early. Low sample purity, low abundance, or degraded material can reduce interpretability in both DNA and protein workflows. For de novo analysis, insufficient analyte amount or heavy contamination can shorten usable sequence-tag coverage.
Unexpected findings often need confirmation by replication, alternate digestion, additional primers, or another orthogonal assay. Mixed-template DNA, carryover, keratin contamination, co-purifying proteins, or sample-handling artifacts can also complicate interpretation. Apparent novelty should be checked against these routine explanations before it is treated as a sequence-level discovery.
A confirmed coding sequence does not prove the final mature protein form. Conversely, de novo sequencing may support a candidate analyte sequence while still leaving uncertainty at specific residues or PTM sites. That is why the better next step should be chosen by the unresolved question, not by repeating the same method category.
FAQ
Can Sanger sequencing still be the right choice if I already have access to NGS?
Yes. If the question is limited to a short, known region and the sample is essentially a single template, Sanger sequencing is often the cleaner way to confirm that specific change. NGS can add complexity without adding decision value for that narrow task.
What does protein inference miss that direct LC-MS/MS can address?
Protein inference starts from a coding sequence or transcript and predicts the translated product. It may miss isoform differences, signal peptide cleavage, truncations, or PTMs that change the analyte actually present. LC-MS/MS examines peptides derived from the sample itself, so it can test whether the measured molecule fits that prediction.
When do unmatched spectra become a decision signal rather than a simple data-quality issue?
They become more meaningful when they persist after routine cleanup, sensible search settings, and contaminant review. If unmatched spectra remain alongside an unexpected intact mass or partial sequence inconsistency, the issue may reflect true sequence novelty, processing, or modification rather than database tuning alone.
Is de novo peptide sequencing only for completely unknown samples?
No. It is also useful when a sample is only partly known but the observed data do not fit the expected translation. Common examples include recombinant products with unexplained mass shifts, proteins from non-model organisms, and PTM-rich analytes with weak database support.
Final Decision Guidance
Choose Sanger sequencing for narrow template confirmation. Choose NGS for broader discovery, depth, or mixed-template analysis. Move toward de novo peptide sequencing or de novo protein sequencing when the decision depends on the peptide or protein form present in the sample rather than the template that may have produced it. For projects in that transition zone, pair the technical summary with the real sample context, then contact us to evaluate your project with MtoZ Biolabs around LC-MS/MS data review, sequence-confidence limits, and follow-up validation options.
Related Services
Main Service |
Supporting Service |
Validation Service |
Alternative Service |
How to order?

