





NGS Antibody Sequencing: When Repertoire Data Helps and When It Does Not Give You the Final Clone Sequence
- comparing pre- and post-selection binder pools
- ranking candidate clonotypes for follow-up
- checking whether selection is converging on a small number of sequence families
- examining CDR3 usage and V(D)J recombination patterns
- deciding whether a pool is diverse enough to justify deeper recovery work
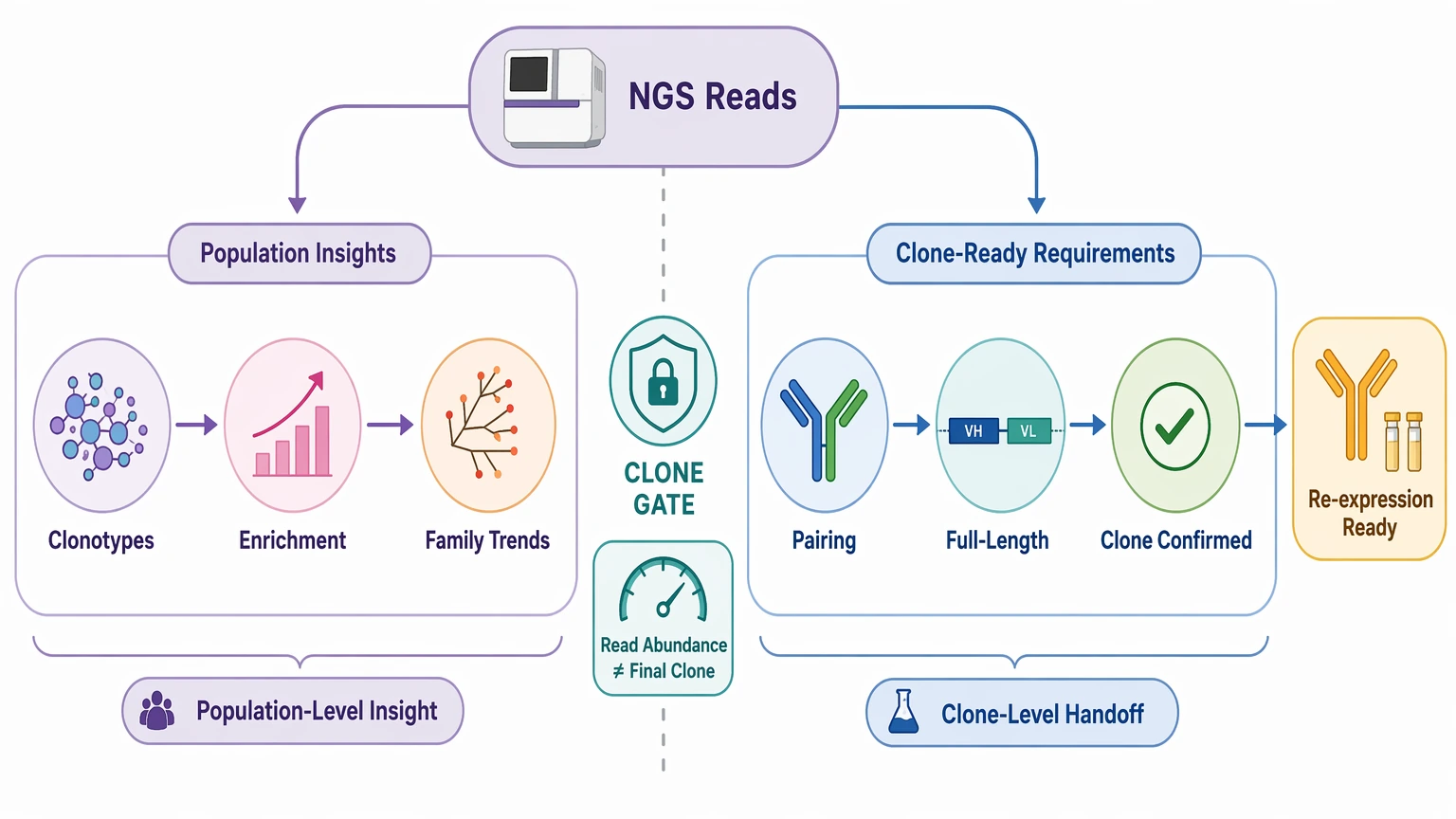
Standard NGS antibody sequencing is most useful when the goal is to describe a population of antibody sequences, not to hand off a single final clone that is ready for re-expression. In practice, bulk repertoire workflows work well for antibody repertoire sequencing tasks such as clonotype tracking, V(D)J recombination profiling, CDR3 enrichment analysis, germline assignment, and somatic hypermutation pattern review. They are much less decisive when a team needs one confirmed VH and VL pair with enough sequence certainty for construct design.
That difference usually drives the real workflow choice. Repertoire data can shrink a binder pool, highlight enriched sequence families, and show whether selection is converging. What it often does not settle on its own is native heavy chain / light chain pairing, full-length variable-region recovery, or clone sequence confirmation. If the endpoint is recombinant re-expression, the sequencing plan should be built around that deliverable, not around read count alone.
Where This Decision Appears in Real Projects
This question usually comes up after immunization, display selection, hybridoma generation, or B-cell recovery, when a discovery team has sequence-accessible material but still does not have an executable antibody sequence. The input may be bulk B cells, an enriched binder pool, hybridoma-associated RNA, or mixed nucleic acid from a selection output.
At that point, standard NGS antibody sequencing often looks appealing because it surveys many rearrangements in one run. The problem starts when abundant repertoire reads are treated as though they already define the final functional antibody. A dominant heavy-chain clonotype can still have more than one plausible light-chain partner. A strong CDR3 signal can still come from incomplete variable-region coverage. When those limits are overlooked, teams may synthesize the wrong chain combination or move forward with a sequence inferred from pooled data rather than directly recovered.
The Four Comparison Dimensions That Usually Decide the Workflow
1. Clone-resolution capability
Some methods describe a population. Others recover candidate-level sequences. A smaller set of approaches can support exact sequence-level output. If the project only needs enrichment analysis or lineage trends, bulk repertoire sequencing may be enough. If the project needs one confirmed antibody sequence, clone-resolution capability becomes the first filter.
2. Heavy chain / light chain pairing
Pairing is often the main line between useful repertoire data and a final clone handoff. Many bulk repertoire sequencing workflows do not preserve native heavy chain / light chain pairing. Because of that, a dominant heavy chain does not identify the correct light chain by itself, and the reverse is also true.
3. Region coverage and recovery readiness
Short-read antibody repertoire sequencing may recover CDR3 and much of the variable region, but that still does not always mean full-length variable-region recovery. Primer placement, template quality, and read architecture can leave uncertainty near transcript ends or in regions needed for final construct design. Recovery readiness comes down to whether the output is complete enough for gene synthesis and recombinant re-expression.
4. Sample fit and validation needs
Method choice should match the biological source. Bulk repertoires, display-derived binder pools, hybridoma material, and purified antibody do not create the same decision path. The plan also has to account for any follow-up confirmation that may still be needed, such as RACE, hybridoma sequencing, single-cell sequencing, or protein-level sequencing.
Comparison of the Main Options
The methods below are not interchangeable. They answer different project questions and work at different levels of biological resolution.
| Method | Typical input | Clone-resolution capability | Heavy chain / light chain pairing | Region coverage | Best use and main limitation |
|---|---|---|---|---|---|
| Bulk repertoire sequencing | Bulk B cells, mixed binder pool, display output | Population-level to clonotype-level | Usually absent or inferred | Partial to near-full variable region, workflow-dependent | Diversity profiling, enrichment analysis, candidate narrowing; Sequence ambiguity often remains for final clone confirmation |
| Single-cell sequencing | Individual B cells or sorted cells | Candidate-level to exact paired sequence | Preserved at the cell level | Often supports paired VH and VL recovery | Native pairing recovery from viable cellular input; Requires suitable cells and cell handling |
| RACE | RNA from enriched cells or a clone-associated source | Targeted transcript recovery | Depends on input context | Strong for extending partial variable-region reads | Clarifying transcript ends after NGS prioritization; Less effective for highly mixed populations without candidate focus |
| Hybridoma sequencing | Hybridoma-associated nucleic acid | More clone-centered than bulk repertoire data | Often more tractable than pooled input | Good variable-region recovery potential | Sequence recovery from monoclonal sources; Aberrant transcripts and transcript heterogeneity can remain |
| Protein-level antibody sequencing | Purified antibody without usable nucleic acid | Molecule-specific confirmation route | Inferred from protein evidence, workflow-dependent | Peptide-based sequence reconstruction | Samples lacking RNA or cells; Some positions may still require confirmation logic |
When Bulk Repertoire Sequencing Adds Real Value
Bulk repertoire sequencing is most informative when the question is about how a sequence population behaves. It can support enrichment analysis across selection rounds, identify expanding clonotype families, map germline assignment patterns, and show how somatic hypermutation accumulates within dominant lineages.
That is useful in several common discovery settings:
Used this way, repertoire data acts as a front-end discovery filter. It tells teams where to focus recovery effort rather than pretending to replace recovery.
Why Repertoire Data Often Does Not Deliver the Final Clone Sequence
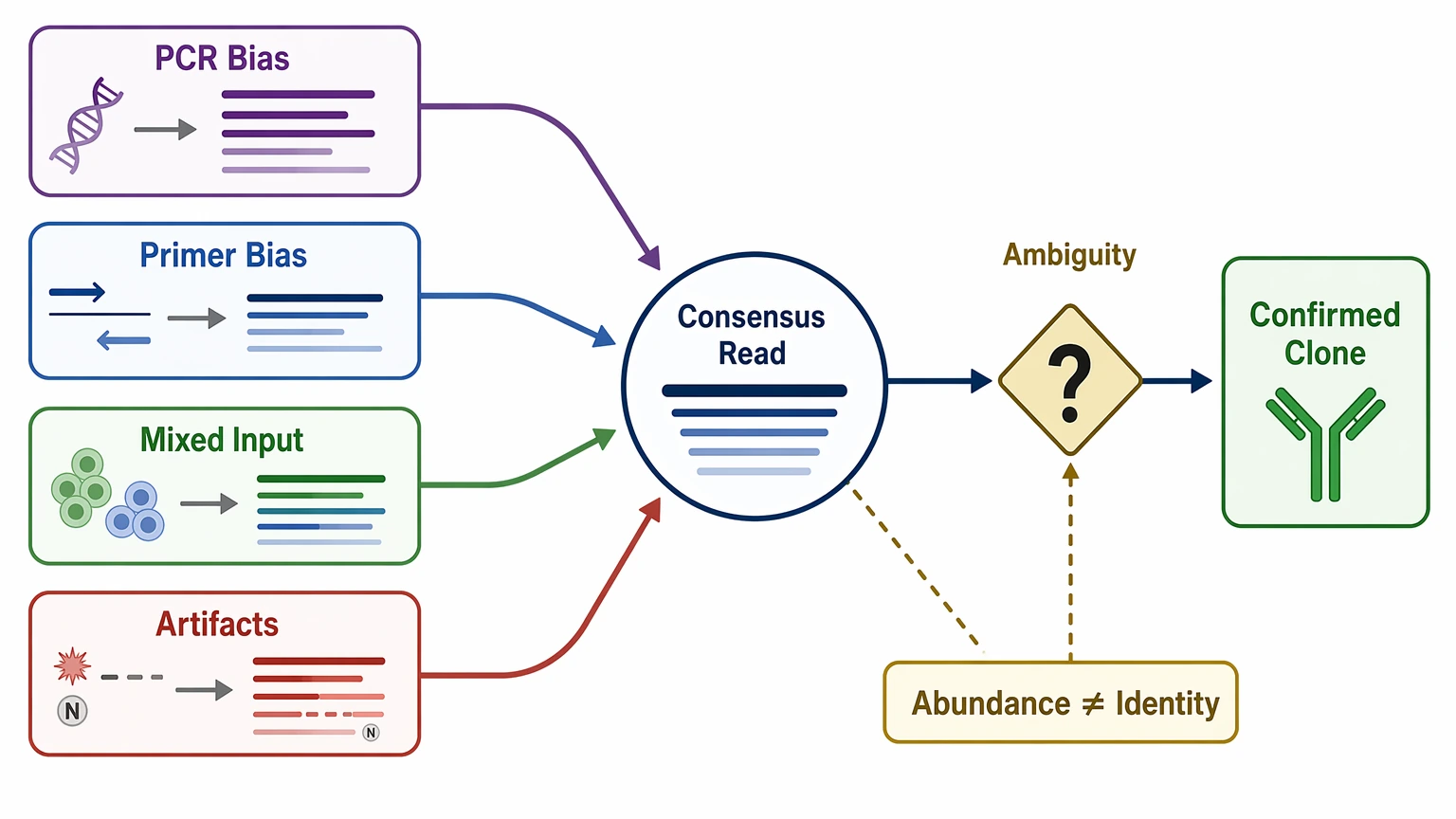
The most common limitation is missing heavy chain / light chain pairing. In bulk repertoire sequencing, heavy-chain and light-chain reads are often measured as separate populations. Even when both chains show dominant clonotypes, the native pair can still be uncertain.
Coverage is the second limitation. Some workflows emphasize CDR3 or partial variable-region segments, which can support clonotype identification but still stop short of full-length variable-region recovery. That gap matters when the project needs exact sequence boundaries for recombinant re-expression.
Bias and ambiguity matter too. PCR amplification, primer bias, mixed-population input, and sequencing artifacts can distort apparent abundance. A consensus sequence may reflect the most frequent read pattern in the dataset, but that is not the same thing as confirmed biological identity. Put simply, repertoire data can be highly informative and still fall short of a final sequence handoff.
When to Move Beyond Bulk Repertoire Sequencing
Choose single-cell sequencing when pairing is the blocking issue
If viable cells are available and the project needs exact paired VH and VL information, single-cell sequencing is usually closer to the final deliverable than bulk repertoire sequencing. Its main advantage is native chain linkage at the cell level, which cuts down the number of speculative pair combinations that would otherwise need testing.
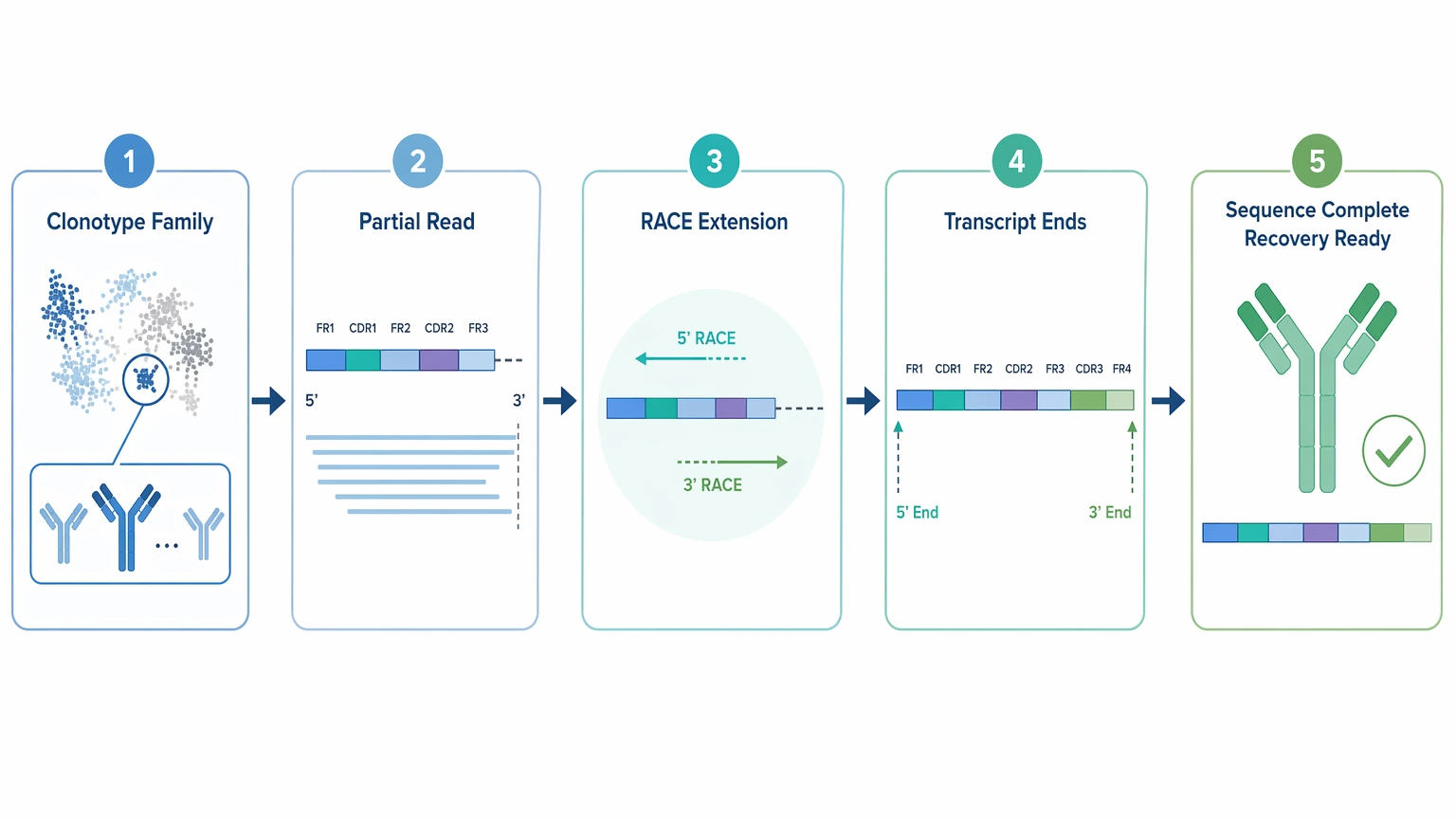
Use RACE when NGS has narrowed the search but not completed the sequence
RACE is especially useful after antibody repertoire sequencing has already identified a likely clonotype family. It can extend partial reads, define transcript boundaries, and improve recovery readiness when the remaining problem is incomplete variable-region coverage rather than broad population complexity.
Prefer hybridoma sequencing for clone-centered material
When the source is a credible hybridoma, hybridoma sequencing is usually a better fit than bulk repertoire sequencing for final sequence recovery. The biological input is narrower, so the workflow starts closer to monoclonal identity. Even then, aberrant chains or transcript heterogeneity can still create sequence ambiguity, so clone sequence confirmation still matters.
Move to protein-level sequencing when nucleic acid is no longer available
If the only remaining material is purified antibody, NGS antibody sequencing is no longer the lead option. Protein-level sequencing becomes the more relevant route because the recovery problem has shifted from V(D)J-based nucleic-acid analysis to peptide-based sequence reconstruction.
If your team is deciding between these routes before committing samples, MtoZ Biolabs can evaluate your project around sample origin, pairing requirements, current repertoire data, and the expected sequence handoff so the next recovery workflow matches the actual development goal.
A Practical Triage Framework
A short set of project questions will usually show whether repertoire data is enough or whether a recovery-oriented method is the better next step.
| Project question | If yes | If no |
|---|---|---|
| Is the main goal repertoire data, enrichment analysis, or clonotype ranking? | Start with bulk repertoire sequencing | Move to the next question |
| Is the endpoint an exact paired sequence for recombinant re-expression? | Prefer pairing-aware or clone-focused recovery methods | Bulk repertoire sequencing may be sufficient |
| Do you still have viable cells or clone-associated RNA? | Single-cell sequencing, RACE, or hybridoma sequencing remain feasible | Consider protein-level sequencing if only purified antibody remains |
| Is sequence ambiguity mainly caused by missing heavy chain / light chain pairing? | Avoid relying on bulk repertoire sequencing alone | Bulk data may still support prioritization |
What a Good Handoff Looks Like
A useful sequencing plan should define the handoff before the experiment starts. For some teams, the handoff is a repertoire report with clonotype and enrichment analysis. For others, it is a consensus sequence set for targeted follow-up. In later-stage discovery, the handoff may need to be a paired, synthesis-ready variable-region sequence with clear clone sequence confirmation logic.
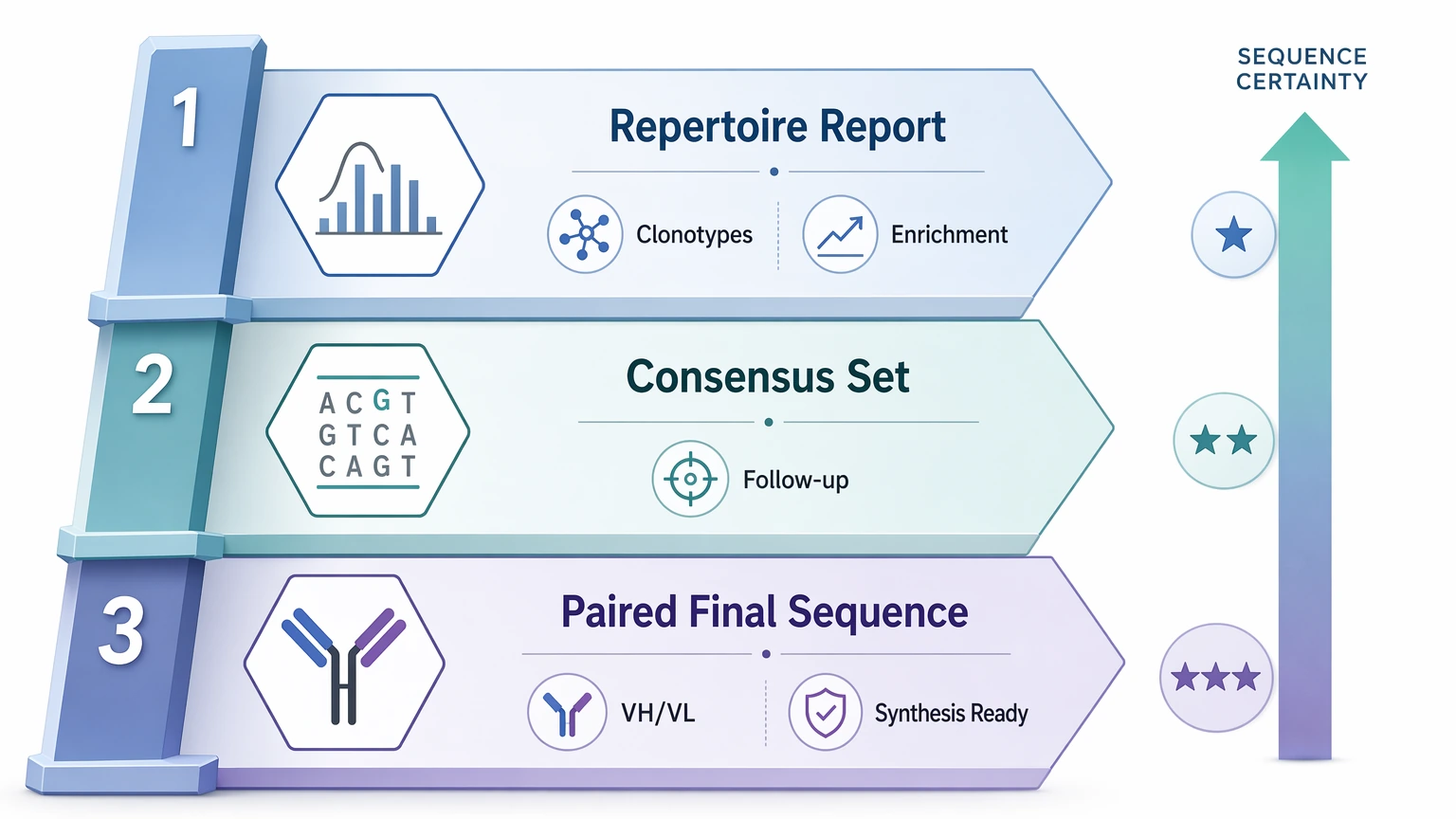
These are different deliverables. Trouble usually starts when a population-level dataset is asked to answer a clone-level question after the fact. A better approach is to decide early whether the project needs library insight, candidate narrowing, or final recovery.
Conclusion
The practical boundary is straightforward: NGS antibody sequencing is often very useful for repertoire structure, enrichment analysis, and candidate prioritization, but standard bulk repertoire workflows do not automatically produce the final paired antibody sequence needed for recombinant re-expression. Teams working with bulk B-cell repertoires, display-derived binder pools, hybridoma-associated material, or purified antibody should choose the workflow based on the required sequence certainty, not on sequencing breadth alone. If you need a project-specific recommendation, contact MtoZ Biolabs to submit your requirements and evaluate your project around sample type, desired deliverable, current repertoire data, and the next confirmation step.
FAQ
Can bulk repertoire sequencing recover the final clone if one clonotype is overwhelmingly dominant?
Sometimes it can narrow the answer to a very small set of candidates, but dominance alone does not prove the native VH and VL pair. The remaining ambiguity is often biological rather than statistical.
Does long-read sequencing automatically solve the clone confirmation problem?
Not automatically. Longer reads can improve variable-region continuity, but they do not guarantee correct heavy chain / light chain pairing unless the workflow also preserves linkage or starts from an appropriate biological source.
Should teams sequence heavy and light chains separately if the goal is fast candidate ranking?
That can work for early enrichment analysis, especially when the immediate goal is tracking clonotype trends. It is much less suitable when the same dataset is expected to support direct recombinant re-expression.
What is the biggest planning mistake before starting an antibody sequencing project?
A common mistake is defining the method before defining the handoff. Teams should first decide whether they need repertoire data, candidate narrowing, or a re-expression-ready sequence package.
Can repertoire data still be useful after a final clone has already been recovered?
Yes. It can help explain lineage context, show whether related clonotypes were also enriched, and support retrospective analysis of selection behavior around the confirmed binder.
What information should be gathered before asking for a sequencing recommendation?
Prepare the sample source, whether viable cells or RNA are still available, whether heavy chain / light chain pairing is required, how complete the variable region must be, and whether the final output must support recombinant re-expression or only candidate prioritization.
How to order?

