





How to Determine Amino Acids from a Mass Spectrum and Locate Modification Sites?
- MS/MS b- and y-ion series show sequential residue masses that map to amino acid compositions when interpreted with a residue mass table.
- Unexpected mass differences between expected residue increments often indicate PTMs such as phosphorylation, acetylation, or oxidation.
- Modification site localization improves when complementary ion series, high-resolution MS, and targeted PTM workflows are used together.
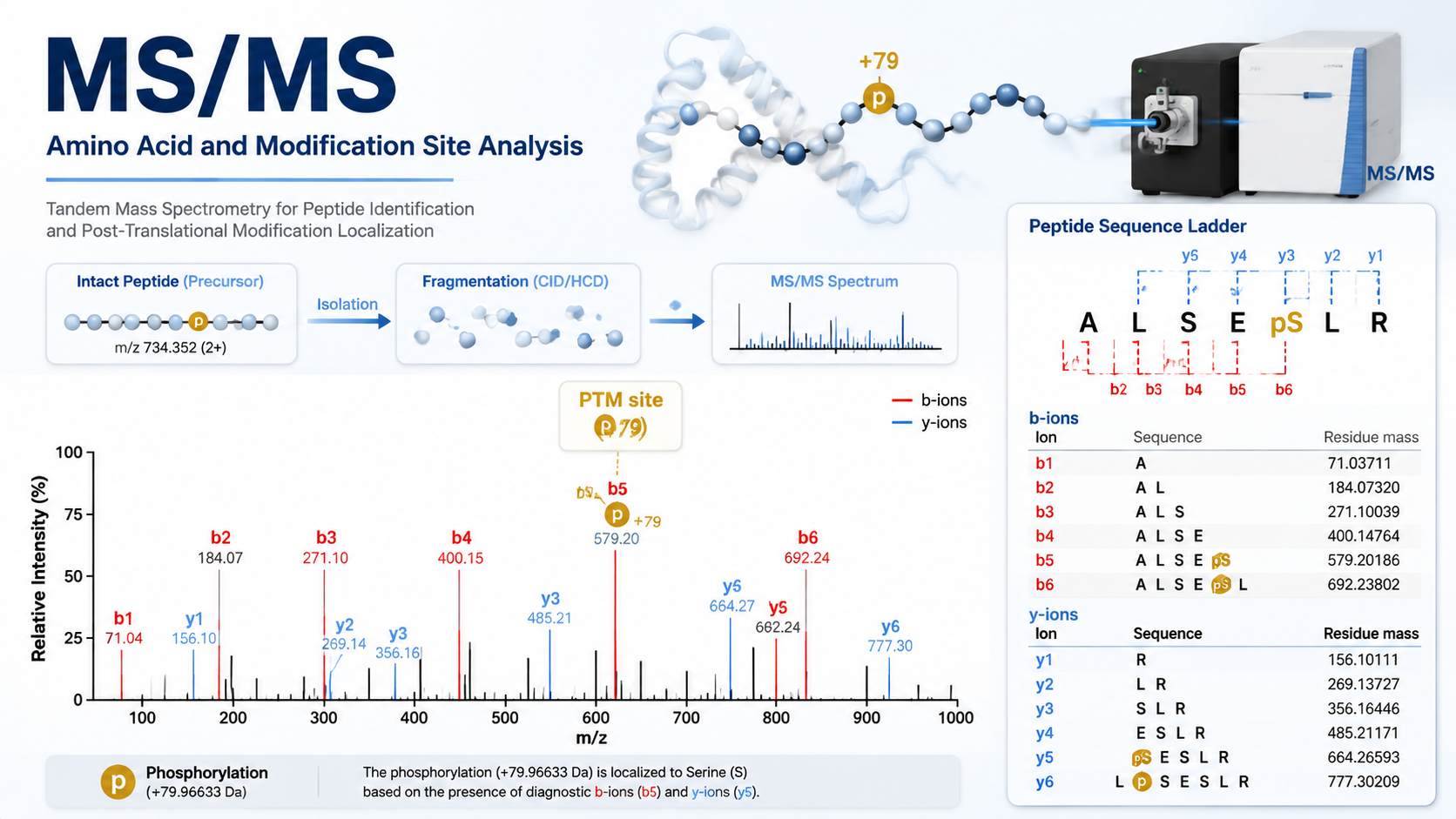
Peptide tandem mass spectra (MS/MS) report fragment ion masses that reflect amino acid composition and modification state. By reading peak series, calculating residue mass differences, and comparing observed shifts to known modification masses, you can infer which amino acids are present and where a post-translational modification (PTM) sits along the sequence.
Key Takeaways
What MS/MS Reveals about Amino Acids?
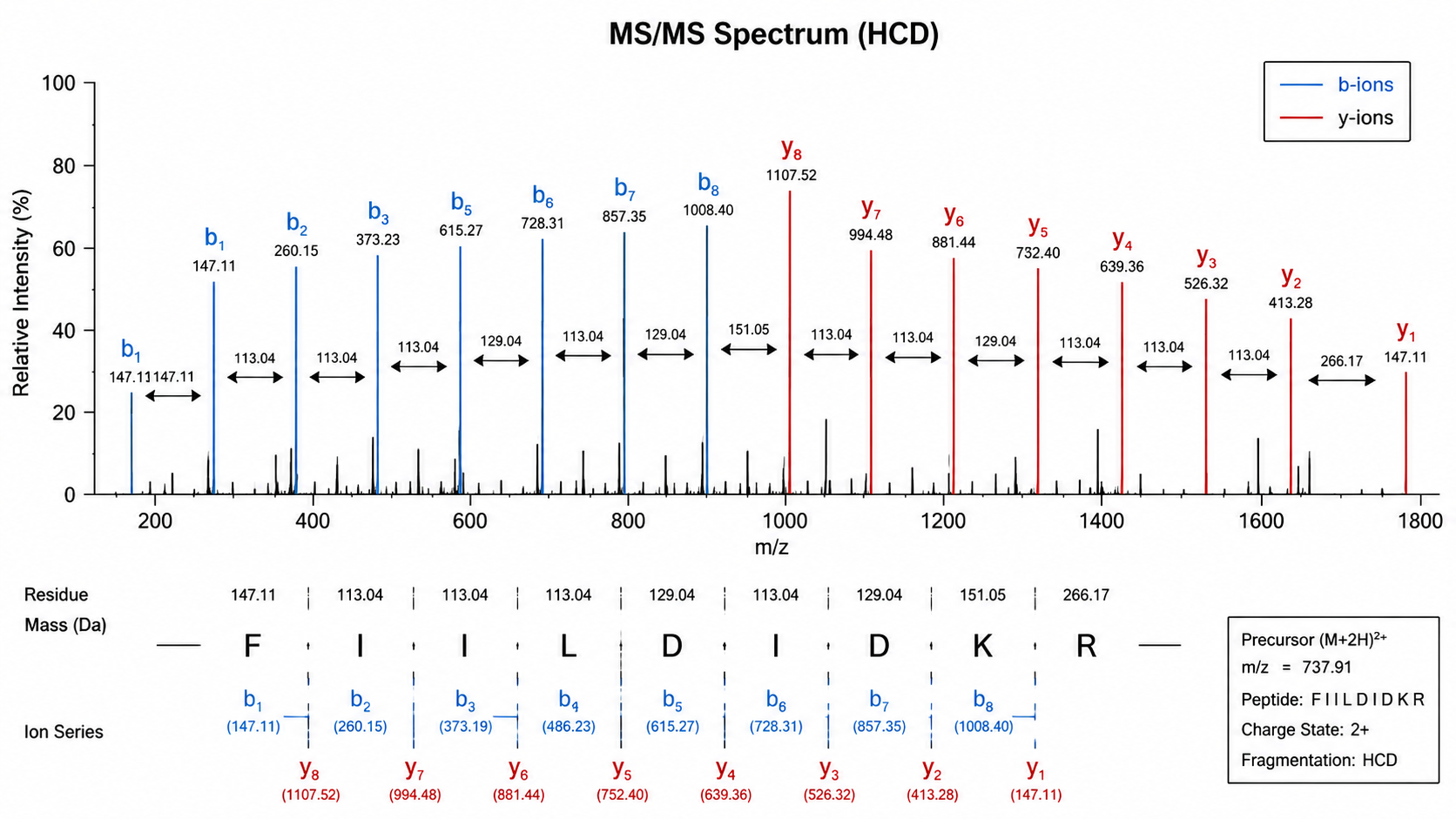
In bottom-up proteomics, proteins are digested into peptides, then fragmented in the mass spectrometer. Each fragment peak corresponds to an ion with a specific mass-to-charge ratio (m/z). For a given peptide, consecutive peaks in a b-ion or y-ion series differ by the mass of one amino acid residue.
Related Services
De Novo Peptide Sequencing Services
Mass Spectrometry-Based Protein Sequencing Service
Edman Based Protein Sequencing Service
Amino Acids Composition Analysis Service
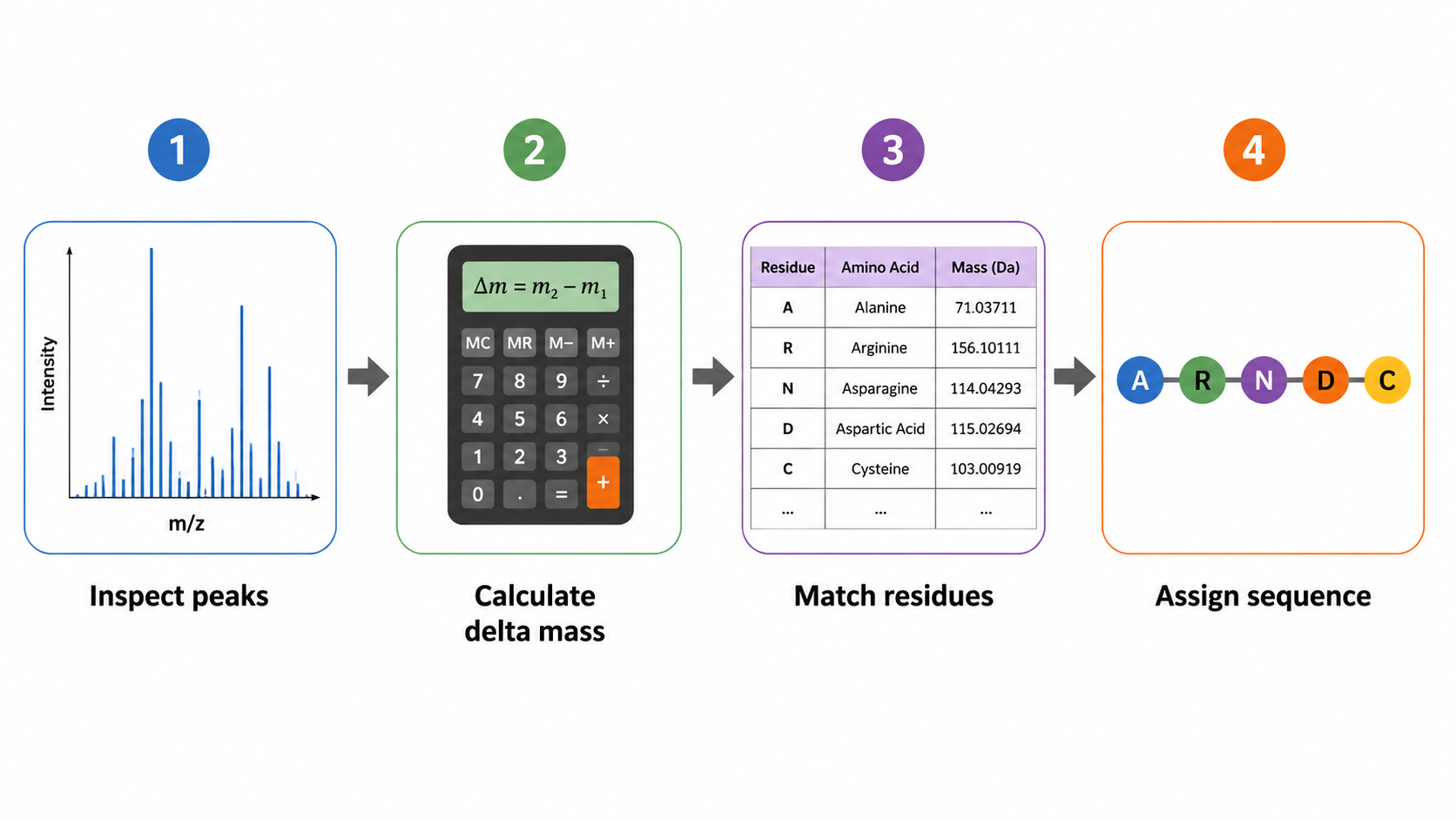
Assign Amino Acids from MS/MS
1. Examine Fragment Ion Peaks
Identify major b-ions (N-terminal fragments) and y-ions (C-terminal fragments). High-abundance peaks that form a regular spacing pattern are the starting point for sequence inference.
2. Calculate Mass Differences
Subtract adjacent peak masses to obtain residue increments and match them to candidate residues. Isoleucine and leucine are isobaric and usually cannot be distinguished by mass alone.
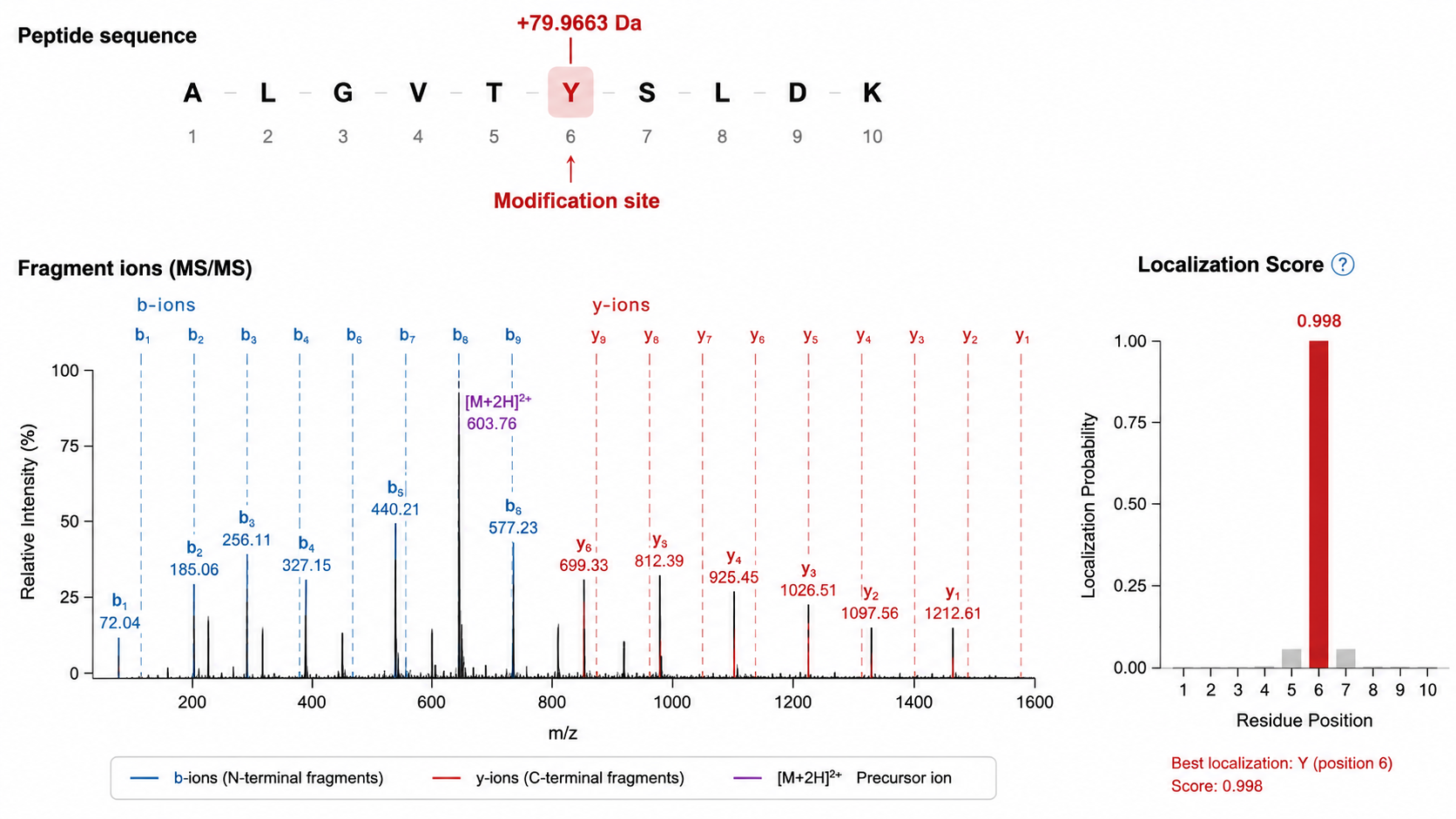
Localize Modification Sites
1. Detect Unexpected Mass Shifts
When a mass difference does not match any standard residue increment, compare the shift to known PTM masses (for example, phosphorylation or acetylation).
2. Localize the Site within the Peptide
Site localization scores compare fragment ions that include versus exclude the modified residue. More fragment ions spanning the modified position yield higher confidence.
Common Pitfalls
| Issue | Why it happens | What to do |
|---|---|---|
| Ambiguous I/L | Isobaric residues | Use orthogonal evidence |
| Incomplete series | Low fragmentation efficiency | Try alternative activation |
| False PTM | Over-search | Control FDR and use enrichment |
FAQ
1. Can you determine every amino acid from one MS/MS spectrum?
Not always. Weak fragmentation, isobaric residues, and labile modifications can leave gaps.
2. How do you tell phosphorylation from a different +80 Da shift?
Use accurate mass, enrichment, and site localization ions together with biological context.
3. When is de novo sequencing needed?
When no database sequence exists or when verifying unexpected peptide sequences.
Conclusion
Reading amino acids from a mass spectrum links fragment ion spacing to residue masses, then tests whether any increment carries a modification shift. With careful interpretation and PTM-aware follow-up, MS/MS supports both sequence assignment and confident modification site localization.
How to order?

