





Bottom-Up MS-Based PTM Analysis Service
There are three commonly used mass spectrometry (MS) strategies in proteomics: top-down, bottom-up, and middle-down approaches. Among them, the bottom-up method is currently the most widely applied strategy. Compared to the top-down approach, the bottom-up strategy offers significant advantages in sensitivity, proteome coverage, and high throughput, making it suitable for studying complex samples and low-abundance modifications. Its principle involves enzymatically digesting proteins into short peptides, which are then analyzed using high-resolution mass spectrometry to accurately identify and quantify post-translational modification (PTM) sites. Bottom-up MS-based PTM analysis is built upon this strategy, combining enzymatic digestion, modification enrichment, and mass spectrometry detection to comprehensively analyze the distribution and dynamics of various PTMs, including phosphorylation, acetylation, methylation, ubiquitination, and glycosylation.
Bottom-up MS-based PTM analysis service is widely used across various research areas, including the investigation of signaling pathway regulation mechanisms, profiling disease-related modifications, evaluating the quality of antibodies and recombinant proteins, identifying drug targets and elucidating their mechanisms of action, as well as monitoring protein aging and stress responses. Its broad applications in both basic research and biopharmaceutical development provide powerful technical support for understanding protein function regulation and disease pathogenesis.
Services at MtoZ Biolabs
Based on advanced high-resolution mass spectrometry platforms and nano-scale liquid chromatography systems, MtoZ Biolabs offers a bottom-up MS-based PTM analysis service focused on high-throughput and accurate profiling of protein post-translational modifications (PTMs). This service involves enzymatic digestion of protein samples to generate peptides, followed by mass spectrometric detection to comprehensively identify and quantify various PTM types, including phosphorylation, acetylation, ubiquitination, and glycosylation. The service provides modification site localization, peptide sequences, spectral data, and relative abundance information. The resulting data can be used to investigate protein functional regulation mechanisms, disease-related modification changes, drug target validation, and biomarker discovery, supporting both basic research and biopharmaceutical development.
Analysis Workflow
1. Protein Extraction and Digestion
Target proteins are extracted from cells, tissues, or purified protein samples, followed by reduction and alkylation treatments. Enzymatic digestion with trypsin or other proteases is then performed to generate peptides suitable for mass spectrometry analysis.
2. Modified Peptide Enrichment
For specific post-translational modifications (e.g., phosphorylation, acetylation), enrichment is carried out using TiO₂, IMAC, or antibody affinity strategies to enhance the detection of low-abundance modified peptides.
3. Mass Spectrometry Detection and Analysis
High-resolution mass spectrometry platforms are used to perform LC-MS/MS analysis and acquire peptide mass data.
4. Data Processing and Results Output
Data processing software is employed for database searching and quantitative analysis. Outputs include modification sites, peptide sequences, annotated spectra, and relative abundance data to support downstream functional studies or mechanistic exploration.
Sample Submission Suggestions
1. Sample Types
Supports a variety of sample types including cells, tissues, serum/plasma, and purified proteins. High-purity protein samples are recommended to avoid interference from lipids, nucleic acids, or contaminating proteins, ensuring efficient digestion and high-quality mass spectrometry data.
2. Buffer Requirements
Avoid using buffers containing high concentrations of salts, SDS, glycerol, EDTA, or other components incompatible with mass spectrometry.
3. Sample Transport
Samples should be stored at –80°C and shipped on dry ice to prevent modification loss or protein degradation due to repeated freeze-thaw cycles. For liquid samples, lyophilized or concentrated forms are recommended to improve stability.
Service Advantages
1. Advanced Analytical Platforms
Powered by high-resolution mass spectrometry systems such as the Orbitrap Fusion Lumos and Q Exactive HF, combined with nano-scale liquid chromatography, enabling highly sensitive and accurate detection and quantification of modifications.
2. High Quantitative Accuracy
Supports multiple quantification strategies including Label-Free and TMT, ensuring precise evaluation of modification level changes across samples—ideal for studying dynamic regulatory mechanisms.
3. Comprehensive Enrichment Strategies
Utilizes a range of enrichment methods such as TiO₂, IMAC, and antibody affinity tailored to specific PTMs, effectively enhancing the detection of low-abundance modified peptides.
4. One-Stop Customized Service
Covers the entire workflow from sample preparation, peptide enrichment, and mass spectrometry analysis to data interpretation, offering efficient and flexible solutions for diverse research and application needs.
Applications
1. Signal Transduction Pathway Research
By identifying modification sites and dynamic changes on key proteins, this analysis helps elucidate intracellular signaling mechanisms and their roles in kinase regulation, receptor activation, and related processes.
2. Disease Mechanism Exploration
Bottom-up MS-based PTM analysis service enables the investigation of post-translational modification (PTM) patterns under disease conditions—such as phosphorylation or ubiquitination—to uncover the molecular mechanisms underlying cancer, neurodegenerative disorders, autoimmune diseases, and more.
3. Biomarker Discovery
Through quantitative analysis of modification site expression differences across sample groups, the service aids in identifying disease-associated PTM events, supporting early clinical diagnosis and personalized therapy strategies.
4. Drug Mechanism Validation
Bottom-up MS-based PTM analysis service can compare PTM levels before and after drug treatment, identify affected target proteins and their modification sites, and support drug target validation and mechanism-of-action studies.
Case Study
1. Comprehensive Analysis of Histone Post-Translational Modifications in Mouse and Human Male Germ Cells
This study employed a bottom-up mass spectrometry-based approach to systematically profile post-translational modifications (PTMs) of histones in male germ cells from mice and humans, aiming to investigate their regulatory roles during spermatogenesis. Testicular cells at various developmental stages were selected as study subjects. Histones were extracted using acid extraction, digested with trypsin and Arg-C, and analyzed via high-resolution LC-MS/MS to detect multiple modifications on H3 and H4, including acetylation, methylation, and phosphorylation. The results revealed significant differences in PTM patterns across species and developmental stages, with certain marks such as H4K91ac and H3K27me3 exhibiting dynamic changes during sperm maturation. The study demonstrated that histone PTMs play crucial roles in chromatin remodeling and epigenetic regulation in male germ cells, and this method provides a robust tool for investigating the mechanisms of spermatogenesis.
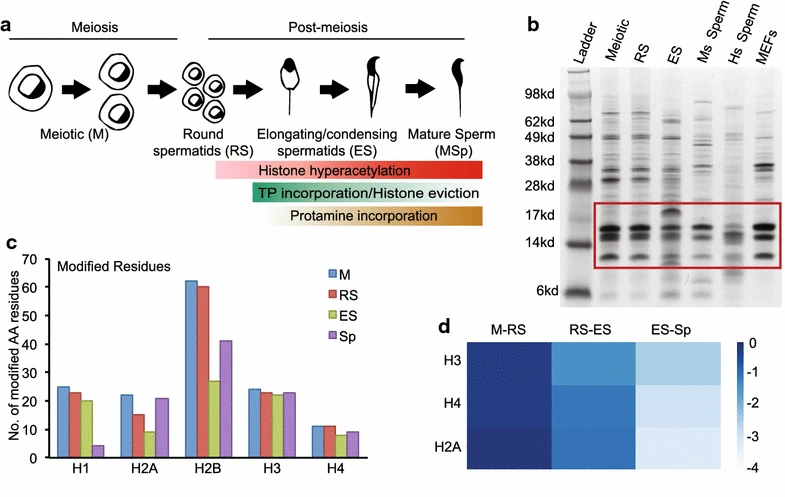
Luense, L J. et al. Epigenetics & Chromatin, 2016.
Figure 1. Schematic Overview and Validation of Histone Post-Translational Modifications Identified during Mouse Spermatogenesis.
2. Bottom-Up Histone Post-translational Modification Analysis using Liquid Chromatography, Trapped Ion Mobility Spectrometry, and Tandem Mass Spectrometry
This study aims to establish a bottom-up mass spectrometry-based analytical strategy that integrates liquid chromatography (LC), trapped ion mobility spectrometry (TIMS), and tandem mass spectrometry (MS/MS) for high-throughput detection of histone post-translational modifications (PTMs). The research focuses on human-derived histones, where chemical derivatization and enzymatic digestion were employed to obtain modified peptides, which were then separated and analyzed using the LC-TIMS-MS/MS platform. The results demonstrate that this method effectively enhances the separation of isomeric modified peptides and accurately identifies multiple PTM sites, such as acetylation and methylation, with high reproducibility and sensitivity. The study concludes that the LC-TIMS-MS/MS technique, combined with a bottom-up approach, enables comprehensive profiling of complex histone PTMs, providing a powerful tool for epigenetic regulation research.
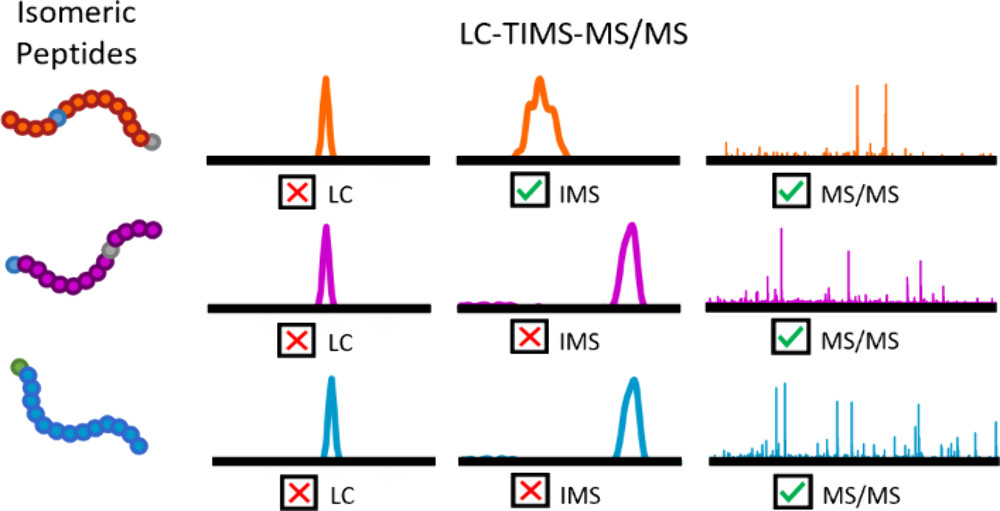
Fuller, C N. et al. Jornal of Proteome Research, 2024.
Figure 2. The Abstract Graphic of this Study.
FAQ
Q1: What Types of Post-Translational Modifications (PTMs) Can this Service Detect?
A1: This service can detect a variety of common PTM types, including phosphorylation, acetylation, methylation, ubiquitination, SUMOylation, and glycosylation. Specific enrichment strategies can be customized based on research needs.
Q2: Does the Service Support Multi-Sample Comparison and Quantitative Analysis?
A2: Yes. Label-Free, TMT, or iTRAQ strategies can be applied to perform relative quantification across multiple sample groups, making it suitable for efficacy evaluation and dynamic monitoring applications.
Q3: Can Low-Abundance Modified Peptides Be Identified?
A3: Yes. By optimizing sample preparation workflows and applying enrichment techniques for modified peptides (such as TiO₂, IMAC, or antibody affinity), combined with high-sensitivity mass spectrometry platforms, the identification rate of low-abundance modifications can be significantly improved.
How to order?

