





LC-MS Histone Modification Detection: PTM Signatures, Workflows, and Quantification
- Histone PTMs often occur on lysine, arginine, serine, threonine, and tyrosine residues.
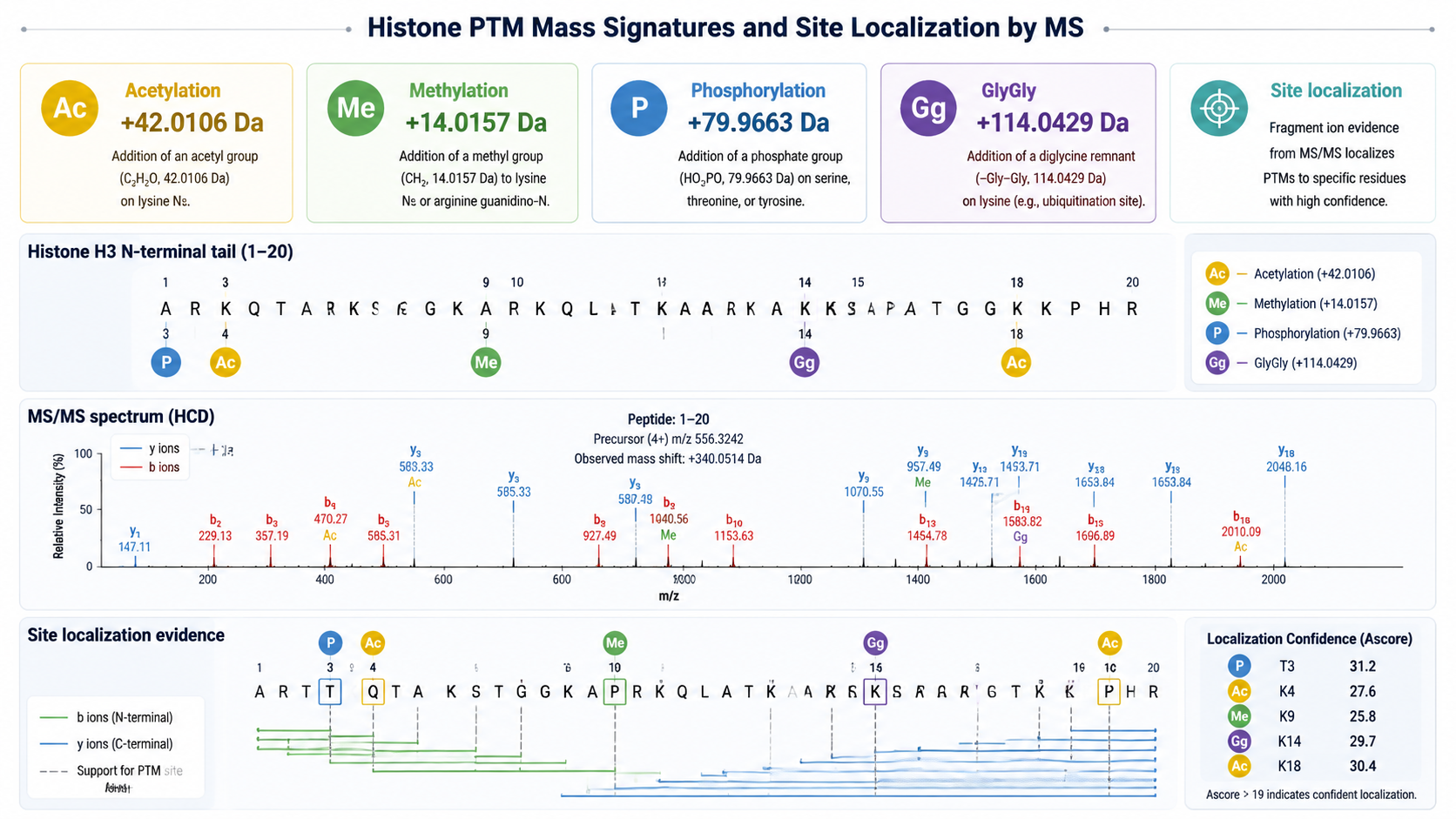
- LC-MS/MS can identify modification type, site, and relative abundance when spectra contain site-determining ions.
- Acetylation and trimethylation have similar nominal mass shifts, so high mass accuracy and fragment evidence are important.
- Bottom-up, middle-down, and top-down MS provide different levels of site and combinatorial PTM information.
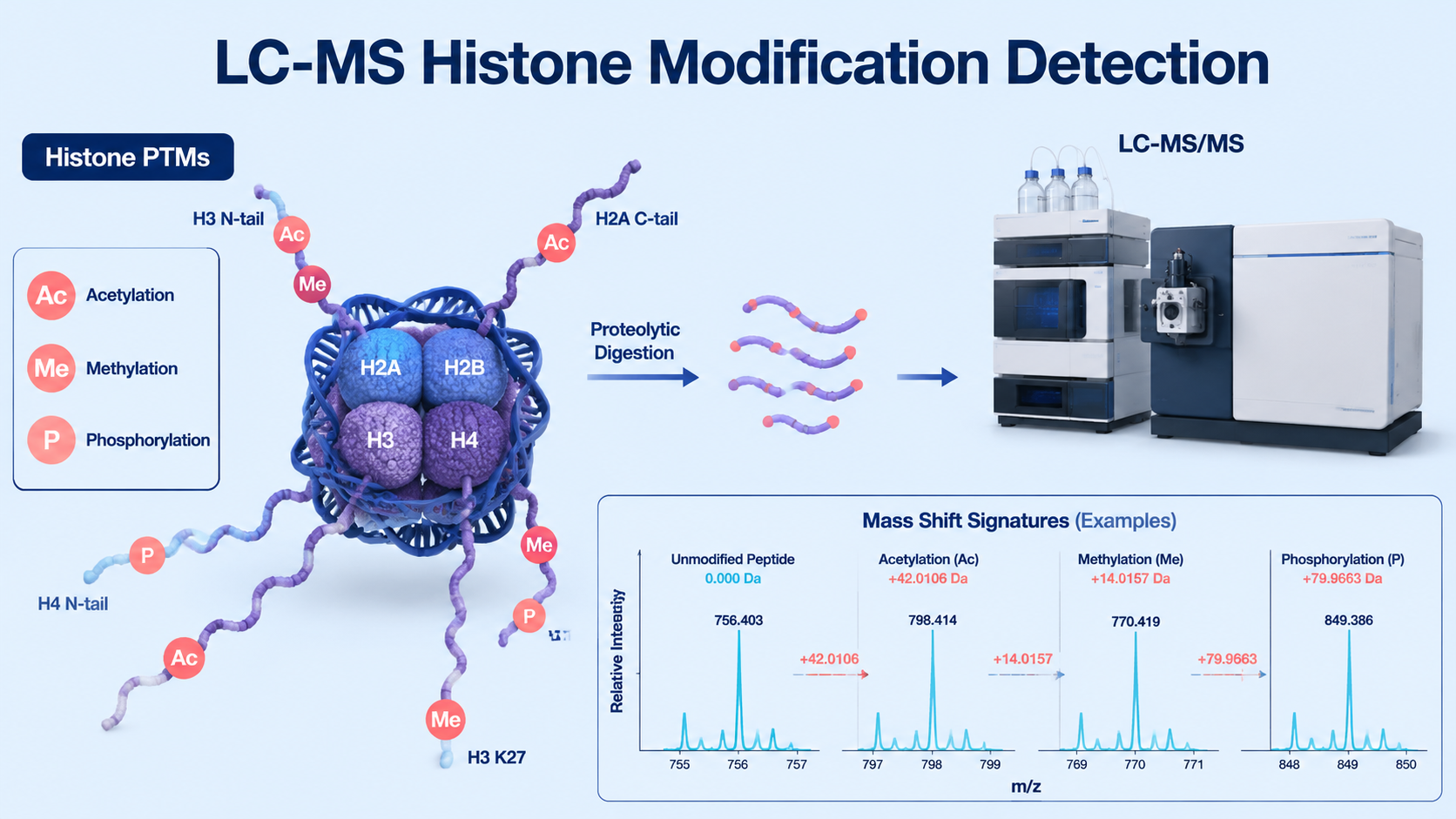
LC-MS histone modification detection identifies and quantifies chemical modifications on histone proteins by separating histone peptides, fragmenting them by tandem mass spectrometry, and matching fragment evidence to specific post-translational modifications (PTMs). Key histone PTM signatures include acetylation (+42.011 Da), mono-methylation (+14.016 Da), di-methylation (+28.031 Da), tri-methylation (+42.047 Da), and phosphorylation (+79.966 Da).
Key Takeaways
What LC-MS Detects in Histone PTM Studies?
Histones carry dense PTM patterns on N-terminal tails and core regions. LC-MS can detect mass shifts on modified peptides, identify the modified residue, and compare abundance across biological conditions. This is useful for studying chromatin regulation, gene expression, DNA damage response, cancer biology, development, and drug response.
Related Services
Histone PTM Identification Service
Histone PTMs Quantitative Analysis Service
Histone PTM Data Analysis Service
Histone Methylation Analysis Service
Common Histone Modification Signatures
| Modification | Common residues | Mass shift | Interpretation note |
|---|---|---|---|
| Acetylation | Lysine, protein N-terminus | +42.011 Da | Often marks active chromatin |
| Mono-methylation | Lysine, arginine | +14.016 Da | Site and methyl state both matter |
| Di-methylation | Lysine, arginine | +28.031 Da | Arginine symmetry may need careful evidence |
| Tri-methylation | Lysine | +42.047 Da | Close to acetylation in nominal mass |
| Phosphorylation | Serine, threonine, tyrosine | +79.966 Da | Neutral loss and site ions support localization |
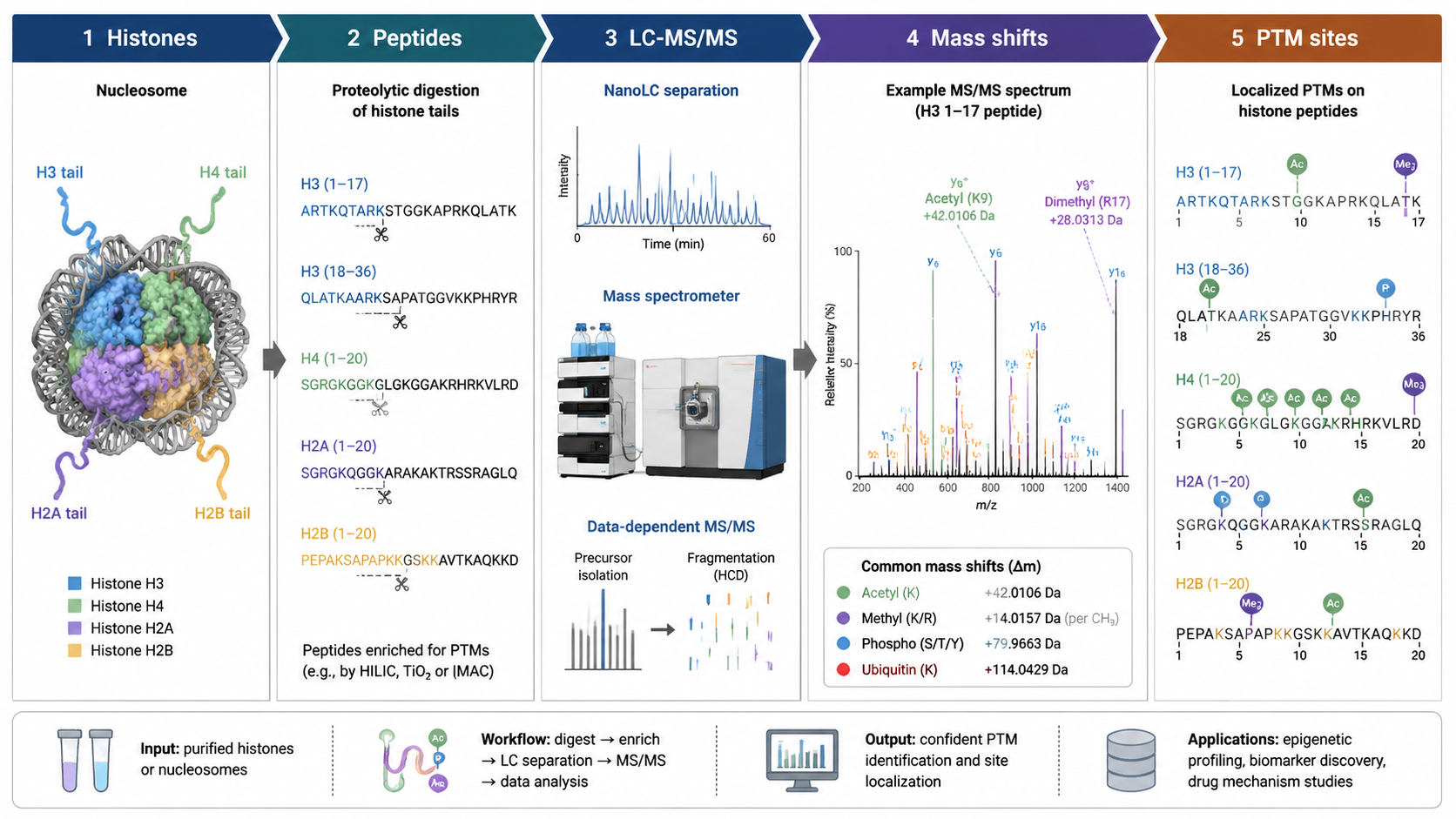
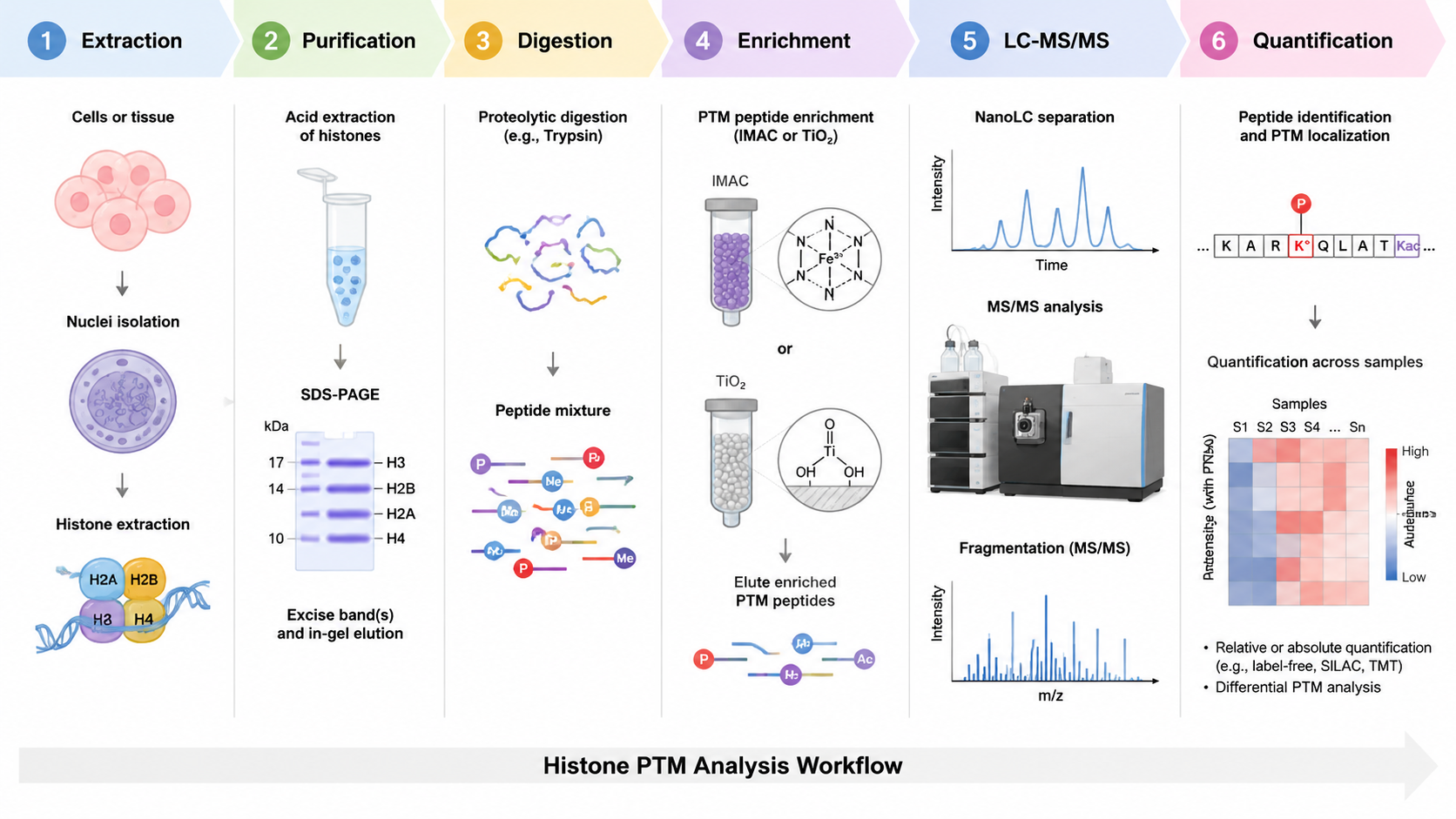
Workflow for LC-MS Histone PTM Detection
Histone analysis begins with nuclear or chromatin-enriched extraction, histone purification, peptide preparation, and LC-MS/MS. For some PTMs, enrichment is useful. Phosphorylated peptides may be enriched by IMAC or TiO2. Histone peptides can be short and highly basic, so derivatization or alternative digestion can improve sequence coverage and quantification.
Site Localization and Combinatorial PTMs
Histone peptides often contain multiple lysines and arginines in short sequence regions. A mass shift alone does not prove the exact site. Site localization requires fragment ions that distinguish candidate residues. When multiple PTMs occur on one peptide, middle-down or top-down strategies may be needed to preserve combinatorial information that bottom-up digestion can lose.
Quantitative Analysis
Quantification can be label-free, isotope-labeled, TMT-based, or targeted. The best approach depends on sample number, expected effect size, instrument platform, and whether the study needs discovery or validation. For key markers, targeted PRM or MRM can confirm selected histone PTM peptides.
FAQ
1. What histone modifications can LC-MS detect?
LC-MS can detect many histone PTMs, including acetylation, methylation, phosphorylation, ubiquitination remnants, lactylation, crotonylation, butyrylation, and other acylations when the method and search settings are designed for them.
2. Why are histone PTMs difficult to analyze?
Histones are highly basic and densely modified. Many peptides contain multiple possible modification sites, and some PTMs have close or overlapping mass shifts.
3. How does LC-MS identify a histone modification site?
LC-MS/MS identifies a site by matching fragment ions to a modified peptide sequence. Site-determining ions are needed to distinguish one possible residue from another.
4. Why can acetylation and trimethylation be confused?
Acetylation and trimethylation have similar nominal mass additions near +42 Da. High-resolution MS and fragment evidence help distinguish them.
Conclusion
LC-MS histone modification detection is powerful because it can connect PTM mass shifts, site localization, and quantitative change in one workflow. Reliable interpretation depends on careful sample preparation, high-resolution MS/MS, correct PTM search settings, and validation of biologically important sites.
How to order?

