





How to Interpret Clonotype Expansion in Deep Sequencing and Human Antibody Repertoire Analysis
- one or two clonotypes occupy an unusually large share of productive reads
- repertoire diversity drops sharply in one batch but not in matched replicates
- a dramatic fold-change comes from a clone with almost no baseline abundance
- V gene usage shifts without a parallel pattern in CDR3 amino acid sequence frequencies
- candidate leads are inferred from heavy chain data without paired heavy/light chain information
- total productive reads
- unique clonotype count
- singleton proportion
- productive versus nonproductive rearrangement ratio
- deduplication approach, including UMI use or non-UMI assumptions
- whether the output contains heavy chain only or paired heavy/light chain information
- Are you tracking an emerging response over time?
- Are you comparing groups for shared enrichment?
- Are you selecting candidates for variable-region recovery?
- Are you deciding whether the dataset is ready for recombinant re-expression?
- exact CDR3 amino acid sequence match
- V gene usage plus J gene usage plus exact CDR3
- similarity-based clustering with a documented threshold
- low-count filtering with and without singleton removal
- top clone fraction or cumulative abundance of top clonotypes
- clonal richness
- evenness
- Shannon entropy
- CDR3 length distribution
- V gene usage and J gene usage skew
- longitudinal fold-change across matched time points
- isotype context, when available
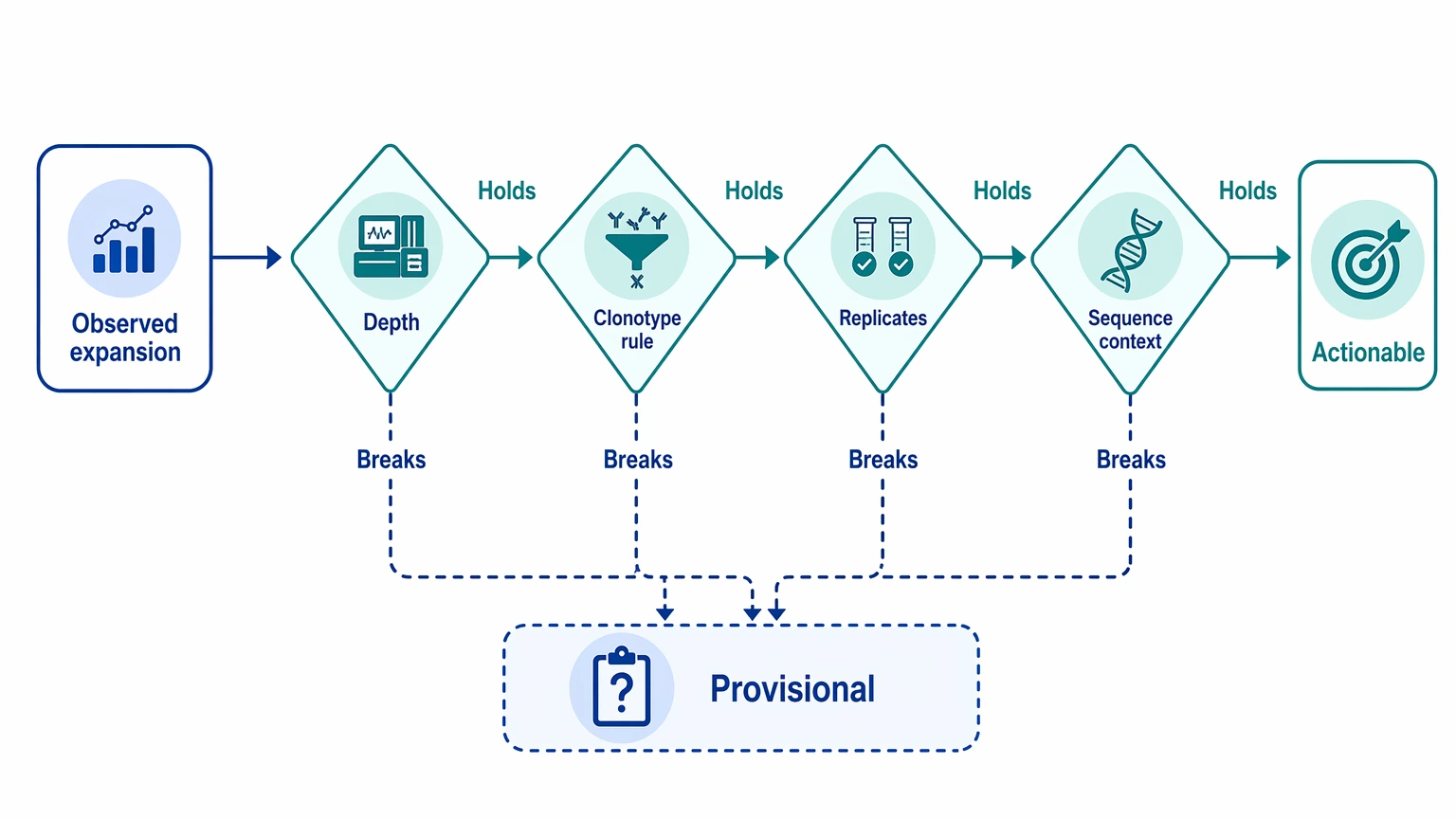
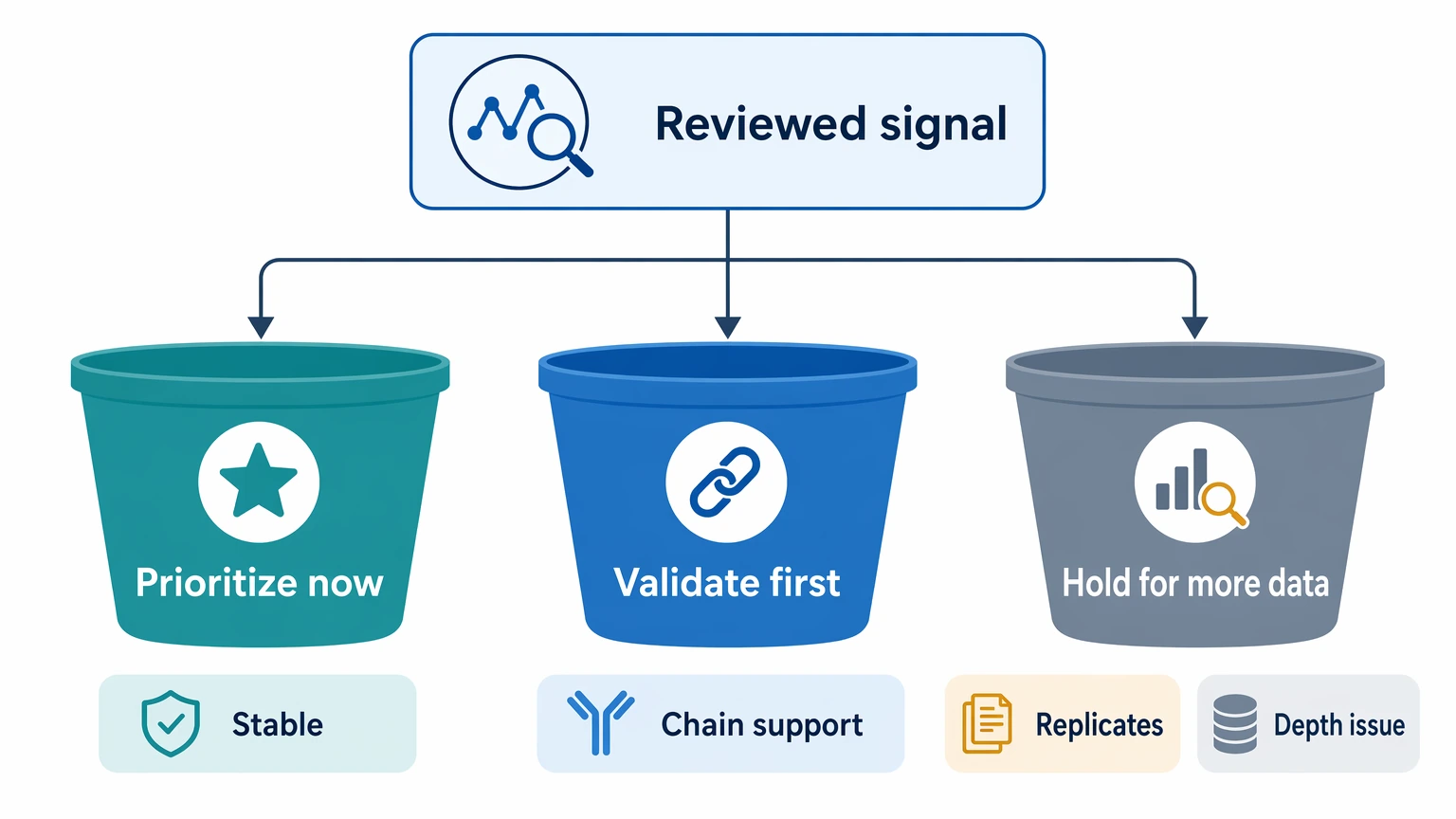
- Prioritize now when clonotype expansion remains stable after depth, filtering, and definition review, and the dataset supports sequence recovery.
- Validate first when the pattern looks plausible but depends on heavy chain abundance alone, weak replicate support, or definition-sensitive clustering.
- Hold for more data when the signal is driven mainly by depth imbalance, low-count instability, or uncertain deduplication.
- stable V(D)J annotation for priority clonotypes
- reproducible clone abundance across replicates or adjacent time points
- full or near-full sequence recoverability rather than partial signatures
- chain context, especially when paired heavy/light chain information is missing
- orthogonal validation plans for the narrowed candidate set
An observed clonotype expansion is worth follow-up only after a structured review. Begin by checking whether the signal still holds after you look at sequencing depth, the clonotype-definition rule, replicate concordance, and sequence context. If one of those checks breaks the pattern, the result can still be useful, but it should stay in the provisional category rather than moving straight into sequence recovery or downstream antibody decisions.
Teams working in deep sequencing and human antibody repertoire analysis often make the same early move: they rank clonotypes by abundance and stop there. That shortcut misses the harder part of the interpretation. A high-frequency clonotype can reflect true clonal expansion, but it can also come from shallow library complexity, deduplication assumptions, over-clustering of related CDR3 sequences, or conclusions drawn from heavy chain data alone. A more dependable readout comes from reading clone abundance together with repertoire diversity, V gene usage, J gene usage, CDR3 amino acid sequence patterns, productive rearrangement ratios, and replicate concordance.
The Interpretation Problem After Antibody Repertoire Sequencing
This issue usually shows up right after the first analysis report arrives. A team sees a rising top clone fraction, a lower Shannon entropy score, or several dominant clonotypes in a post-treatment or post-immunization sample. The pattern looks convincing at first glance, but the practical question is still open: does it point to a biologically meaningful B-cell response, or is it mostly an artifact of sampling depth and analysis settings?
A few warning signs should get immediate attention:
These mistakes affect real project decisions. A translational team may read too much into an antigen-driven response. An antibody discovery group may prioritize variable-region recovery for a signal that does not hold up under threshold testing. A study lead may move toward recombinant re-expression before confirming that the dataset can support full sequence recovery. The point is not to reject expansion signals on principle. The point is to separate signals that remain credible after technical stress testing from signals that do not.
The Four Cause Categories That Explain Most Misreads
For this kind of decision, four categories account for most false confidence in clonotype expansion.
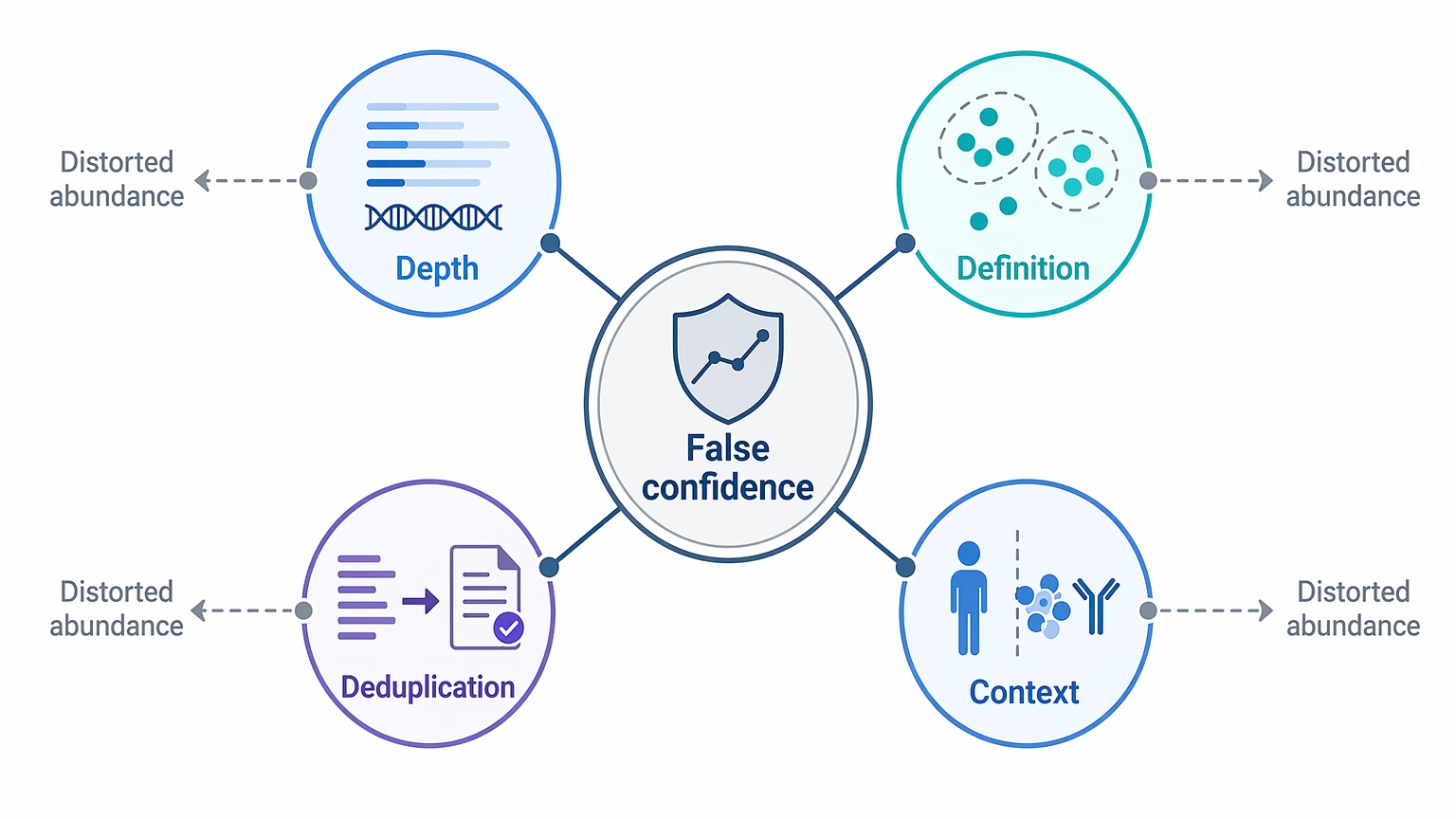
1. Sequencing depth changes what you can see
Sequencing depth affects both the visibility of rare clonotypes and the apparent dominance of abundant ones. In a shallow dataset, the low-frequency tail is undersampled, so the frequency distribution can look narrower than the underlying B-cell receptor repertoire. A low unique clonotype count and a reduced singleton proportion may therefore say more about limited sampling than about real biological focusing.
2. Clonotype definition rules can create or erase expansion
A clonotype is not a fixed biological unit. It is a grouping rule based on V gene usage, J gene usage, CDR3 sequence identity, or similarity thresholds. Exact CDR3 matching, V/J plus exact CDR3 grouping, and similarity-based clustering can all produce different clone abundance estimates. Over-collapsing merges distinct rearrangements; over-splitting separates related members of the same response.
3. Deduplication assumptions can inflate clone abundance
If a library lacks a UMI, or if deduplication relies on assumptions that do not match the library structure, read count can drift away from molecule count. PCR amplification bias can then make clonotype expansion look stronger than it really is. In that setting, an abundant clone may still be real, but its reported frequency may overstate its biological weight.
4. Biological context may still be incomplete
A plausible antigen-driven response usually fits the broader study design. Expansion is easier to defend when it matches time-point logic, class-switched compartments, replicate concordance, and coherent CDR3 sequence features. By contrast, bulk heavy-chain-only datasets can show enriched sequence signatures without establishing a complete antibody identity.
A Data-Interpretation Framework for Clonotype Expansion
This is a data-interpretation problem, so the solution should follow a data-interpretation structure rather than a generic lab workflow. The most useful path is to move from technical sufficiency to biological actionability.
Step 1: Check whether the dataset can support expansion claims
Before interpreting any dominant clone, review the minimum context for each sample:
A simple review table helps here:
| Metric | What to examine | Why it matters |
|---|---|---|
| Sequencing depth | Total productive reads per sample | Low depth can exaggerate dominant ranks |
| Unique clonotype count | Retained clonotypes after filtering | Low complexity can mimic clonal narrowing |
| Singleton proportion | One-count clonotypes | Shows tail sampling and filter sensitivity |
| Deduplication | UMI-based or inferred | Affects whether counts reflect molecules |
| Replicate concordance | Agreement across replicates | Supports technical stability |
If these values are missing or inconsistent, do not jump straight to biological interpretation. At this stage, teams that need a project-level review can submit their requirements to MtoZ Biolabs to evaluate whether the available repertoire output is strong enough for sequence recovery planning or whether more validation should come first.
Step 2: Define the project question before ranking clones
The same expanded clonotype can mean different things in different studies. Clarify the immediate question first:
This distinction matters. A clonotype that rises from a low baseline may be interesting in longitudinal monitoring, yet still be a weak candidate for sequence recovery if coverage is partial or light chain information is missing. By contrast, a stable top clone across replicates from a class-switched compartment may justify faster follow-up even when repertoire diversity changes only modestly.
Step 3: Test the signal under alternative clonotype-definition rules
This is one of the most useful stress tests in human antibody repertoire analysis. Recalculate or review the same dataset under alternative but defensible rules:
You are not trying to find one universal correct rule. You are testing whether the same conclusion survives reasonable analytical choices.
| Pattern after reanalysis | Practical interpretation |
|---|---|
| Stable high rank across rules | More consistent with a real enriched clone family |
| Expansion appears only under permissive clustering | Suggests over-collapsing |
| Fold-change disappears after low-count review | Suggests threshold-driven noise |
| Small rule changes produce different lead clones | Not ready for downstream decisions |
A robust expansion signal should still be recognizable when the grouping assumptions become stricter.
Step 4: Read abundance together with repertoire diversity and sequence context
Abundance alone is rarely enough. Read it beside other metrics:
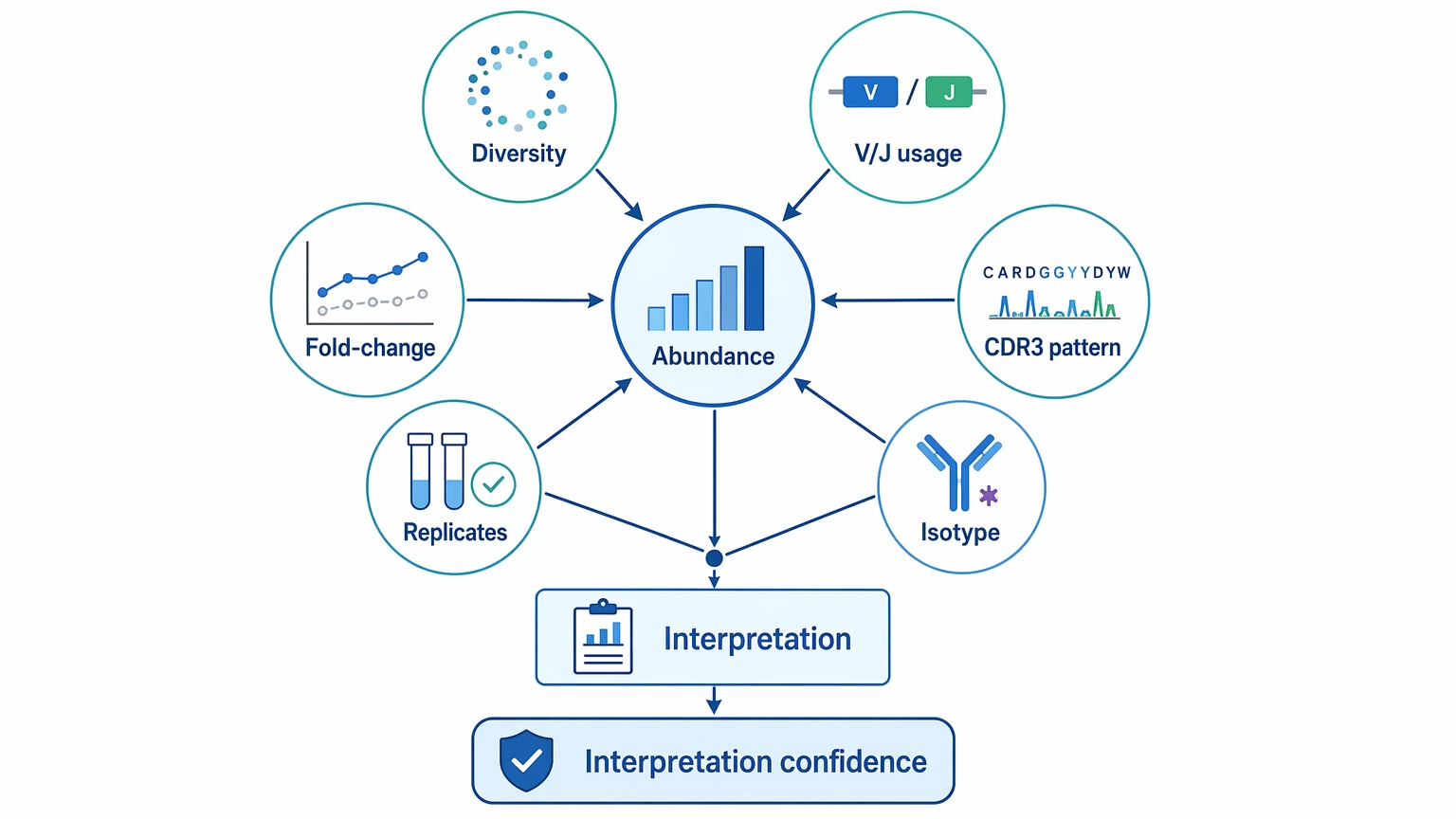
The strongest cases usually combine several aligned observations. If top clone fraction rises while repertoire diversity declines moderately, replicate concordance remains acceptable, and expanded clonotypes share coherent V(D)J recombination context, the case for clonal expansion gets stronger. If diversity drops sharply but the dominant clones move around across replicates, technical bottlenecking becomes the more likely explanation. If V gene usage skews heavily while CDR3 amino acid sequence frequencies remain diffuse, the biological readout is less convincing.
Step 5: Decide whether the signal is actionable
The last question is not simply whether expansion exists. The real question is what the team can do with it next.
Use three decision buckets:
The threshold should be higher when the next step is recombinant re-expression rather than descriptive repertoire reporting. In that setting, the dataset should support variable-region recovery, chain confirmation, and a defensible link between the reported clonotype and a reconstructable antibody sequence.
Common Misread Scenarios
Several patterns lead teams off course again and again.
Shallow library, inflated dominance: A high top clone fraction may reflect low library complexity rather than strong biological selection.
Over-collapsed clonotypes: Similarity-based grouping can merge nearby CDR3 variants into one apparently dominant clonotype.
Heavy-chain over-interpretation: Heavy chain abundance can mark an enriched response, but it does not by itself define a complete antibody candidate.
Fold-change without baseline context: A large ratio from a nearly absent baseline may look dramatic while still representing a low-abundance, unstable signal.
Diversity decline treated as proof: Lower repertoire diversity can accompany clonal expansion, but it does not independently establish an antigen-driven response.
What to Validate Before Moving to Sequence Recovery
When the follow-up goal is variable-region recovery or recombinant re-expression, confirm that the data package can support that transition. Useful checks include:
This is the point where antibody repertoire sequencing stops being only descriptive analysis and starts functioning as sequence decision support. If the dataset cannot support a recoverable sequence path, targeted validation is often the better next move before clone selection.
Conclusion
Clonotype expansion in deep sequencing and human antibody repertoire analysis becomes persuasive when the abundance signal stays stable through depth review, clonotype-definition testing, deduplication review, replicate concordance, and sequence-context checks. That framework fits longitudinal immune studies, translational comparison sets, and antibody discovery programs that need to decide whether a dominant signal is ready for sequence recovery or still needs validation. If your team is at that handoff point, organize the productive-read summary, clonotype-definition rule, replicate structure, and chain-availability details, then contact MtoZ Biolabs to discuss the dataset and submit your requirements for the next technical step.
FAQ
Does a highly expanded clonotype always indicate an antigen-driven response?
No. A strong abundance signal can be consistent with an antigen-driven response, but it can also come from clustering choices, shallow sampling, or amplification skew. The interpretation gets stronger when the signal also matches time-point structure, replicate concordance, and sequence context.
How should I compare clonotype expansion across samples with different sequencing depth?
Start with productive-read depth, unique clonotype count, and singleton proportion before comparing ranks or fold-change. If one sample is much shallower, rare clones may drop out of the denominator and make top clones look artificially stronger.
Is paired heavy/light chain information necessary for follow-up decisions?
Not for every decision. Heavy-chain-only data can still support repertoire-level interpretation and shortlist generation. However, paired heavy/light chain information becomes much more important when the goal is sequence recovery, antibody reconstruction, or recombinant re-expression.
Which diversity metric is most useful when reviewing clonotype expansion?
No single metric carries the whole interpretation. Shannon entropy is useful because it reflects both richness and evenness, but it should be read together with top clone fraction, clonal richness, and the underlying frequency distribution.
When should an expanded clonotype stay off the priority list?
Keep it back when its rank changes sharply after alternative clustering rules, when its fold-change is driven by a near-zero baseline, or when replicate concordance is poor. In those cases, the signal may still deserve monitoring, but it is not yet a strong candidate for immediate recovery work.
How to order?

