





How to Improve Antibody Diversity And Specificity Analysis Optimization in Research Workflows
- If assays disagree but protein sequence evidence is strong, revisit assay design, antigen format, and functional readouts.
- If assays disagree and diversity-informative peptides have weak support, the problem is more likely in unknown peptide or protein identification.
- If both assay and sequence evidence are weak, the workflow needs upstream correction before further interpretation.
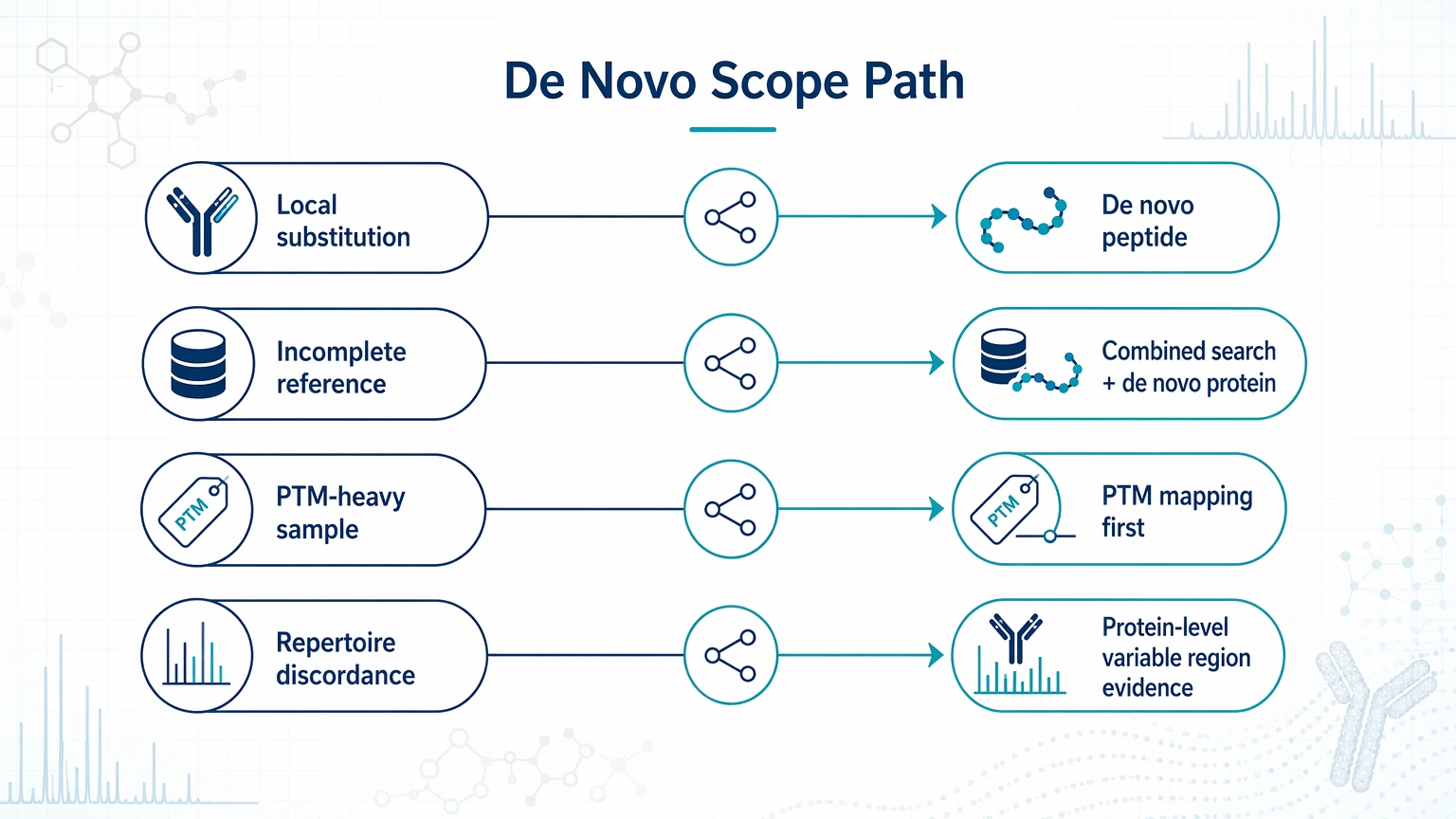
- Local substitution or CDR uncertainty: de novo peptide sequencing
- Incomplete reference for an engineered antibody: combined database search and de novo protein sequencing
- PTM-heavy sample with unexplained mass shifts: PTM mapping first, then de novo review of unresolved regions
- Discordance between repertoire output and expressed binder profile: prioritize protein-level variable region evidence
- Antibody Sequencing Service | Mass spectrometry for unresolved variable-region sequence questions tied to antibody specificity.
- AAE-nanoLC-MS/MS Antibody Coverage Analysis Service when the first issue is insufficient peptide recovery or incomplete variable-region coverage.
- De Novo Protein Sequencing Service when reference gaps extend beyond one peptide or involve partly undocumented constructs.
- LC-MS/MS-Based Targeted Site Validation Service when a candidate finding already exists and needs focused confirmation.
Quick Answer
When antibody diversity and antibody specificity conclusions stay unstable, first determine whether the problem comes from assay behavior or from weak protein-level sequence evidence. Add de novo peptide sequencing or de novo protein sequencing when the unresolved feature sits in a variable region, especially a complementarity-determining region (CDR), and standard database search leaves low-confidence peptide-spectrum matches (PSMs), unmatched spectra, or PTM-related mass shifts that it cannot explain well. If the main weakness is poor sample definition, limited sequence coverage, or uninformative LC-MS/MS spectra, address those issues first.
Use de novo sequencing when the uncertainty is sequence-centered rather than assay-centered, the affected peptide is in a diversity-informative variable region, LC-MS/MS data support residue-level confidence review, and the answer will change clonotype, engineering, or validation decisions.
Do not start with de novo sequencing when the sample is highly mixed, degraded, or contaminated, CDR-containing peptides are not being recovered, spectra are too weak or chimeric for spectral interpretation, or the real next step is a control, repeat run, or orthogonal assay.
Where Antibody Diversity and Specificity Interpretation Breaks Down
Most teams do not lack data. More often, they have several datasets that only partly agree. Repertoire analysis may suggest broad clonotype diversity, while binding data point to a narrower phenotype. A candidate shaped by affinity maturation may show binding behavior that does not match its recorded variable region sequence. LC-MS/MS may also show unexplained mass shifts, sequence variants, or PTM signatures that database search cannot assign with confidence.
In those situations, sequence interpretation often becomes the bottleneck. The issue is not whether antibody diversity exists in principle. It is whether the protein species actually measured support the sequence assumptions behind conclusions about antibody specificity.
A practical split helps:
Why Database Search Alone Often Falls Short
Database search remains useful, but it works best when the reference set closely matches the sample. Antibody workflows often move outside that condition. Somatic hypermutation, engineered substitutions, undocumented constructs, sequence variant accumulation, and mixed molecular forms all reduce agreement between the sample and the reference database.
This gap is usually most obvious in CDR-associated peptides. Those peptides often contain the sequence information that matters most for antibody specificity, yet they are harder to detect and assign. Some are too long after digestion, some fragment poorly, and some only partly resemble known germline references. A nominally acceptable PSM may still rely more on framework region similarity than on direct evidence for the variable region feature under review.
PTMs add another layer of difficulty. Glycosylation, deamidation, and oxidation can shift precursor mass and change fragment behavior. In stressed or heterogeneous antibody preparations, those shifts can mimic or hide a real sequence variant. A de novo sequence path can reduce uncertainty, but it does not automatically prove a complete local sequence or its functional consequence, and Leu/Ile ambiguity remains a known limit for isobaric residues.
Step-by-Step Solution for Data-Interpretation Problems
Step 1: Define the exact unresolved finding
Start with one decision-relevant question rather than a broad request for optimization. Examples include a weakly supported CDR substitution, a mismatch between clonotype assignment and protein evidence, a PTM-bearing peptide that may change specificity interpretation, or an engineered region that lacks a trustworthy reference. This keeps the analysis focused. If the question would not change a project decision, deeper sequence reconstruction may add cost without removing the actual workflow bottleneck.
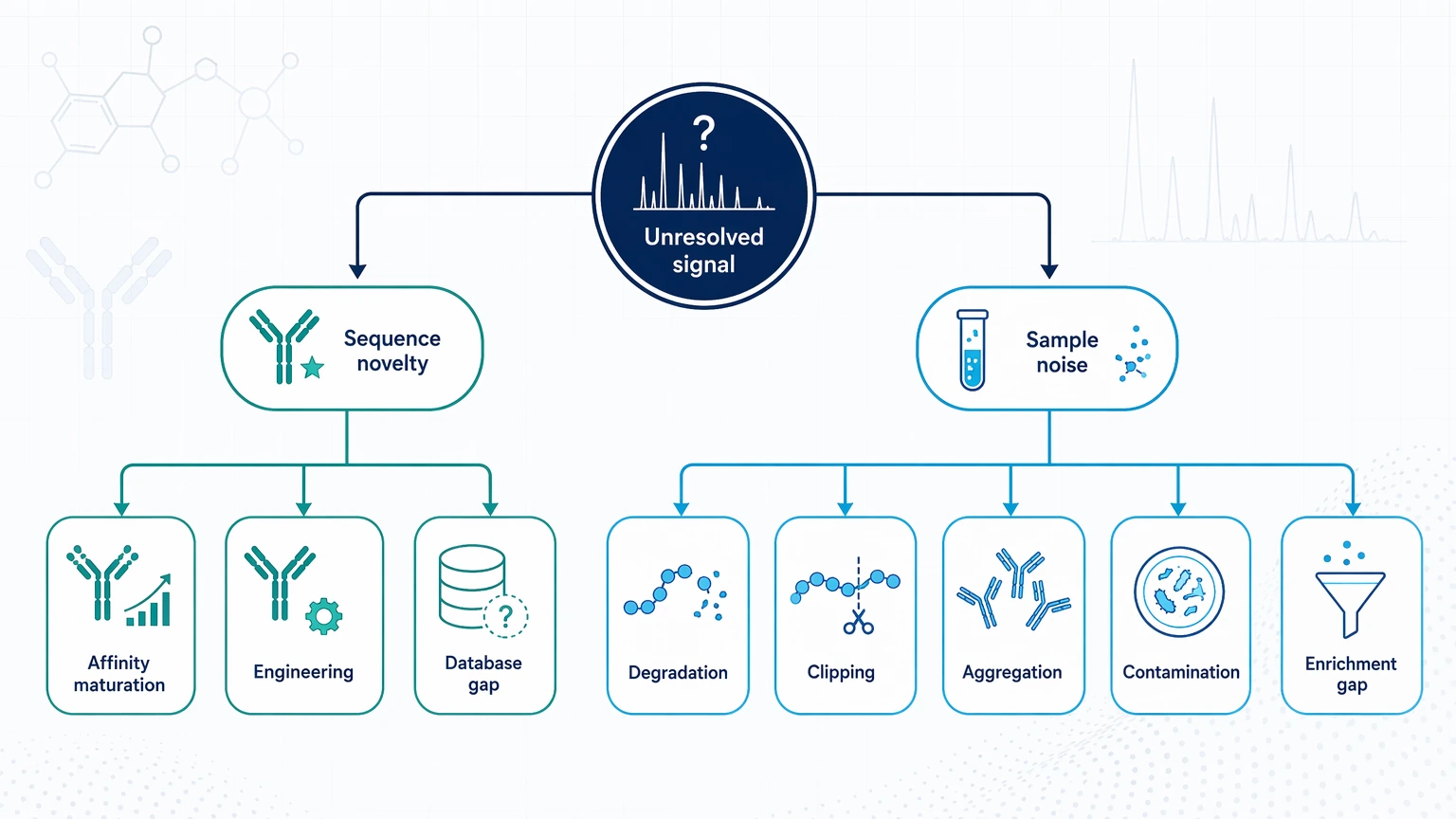
Step 2: Separate sequence novelty from sample or assay noise
Next, ask whether the unexplained signal is more consistent with novelty or with poor material quality. True novelty may involve affinity maturation changes, undocumented engineering, or limited database coverage. Noise is more often tied to degradation, clipping, aggregation, contaminating proteins, or incomplete enrichment. This distinction matters because de novo peptide sequencing works best when the sample still contains interpretable signal. It is less useful when unresolved sample complexity is the main problem.
Step 3: Check whether the LC-MS/MS dataset is actually evaluable
For antibody-related interpretation, the most useful technical checks are fragmentation completeness across diversity-informative peptides, sequence coverage across the variable region rather than only total coverage, precursor isolation quality and spectral cleanliness, peptide uniqueness that separates shared framework peptides from CDR-relevant peptides, and local residue-level confidence rather than a global average score. A dataset can look acceptable in summary and still miss the real decision point if CDR-containing peptides are absent or poorly fragmented.
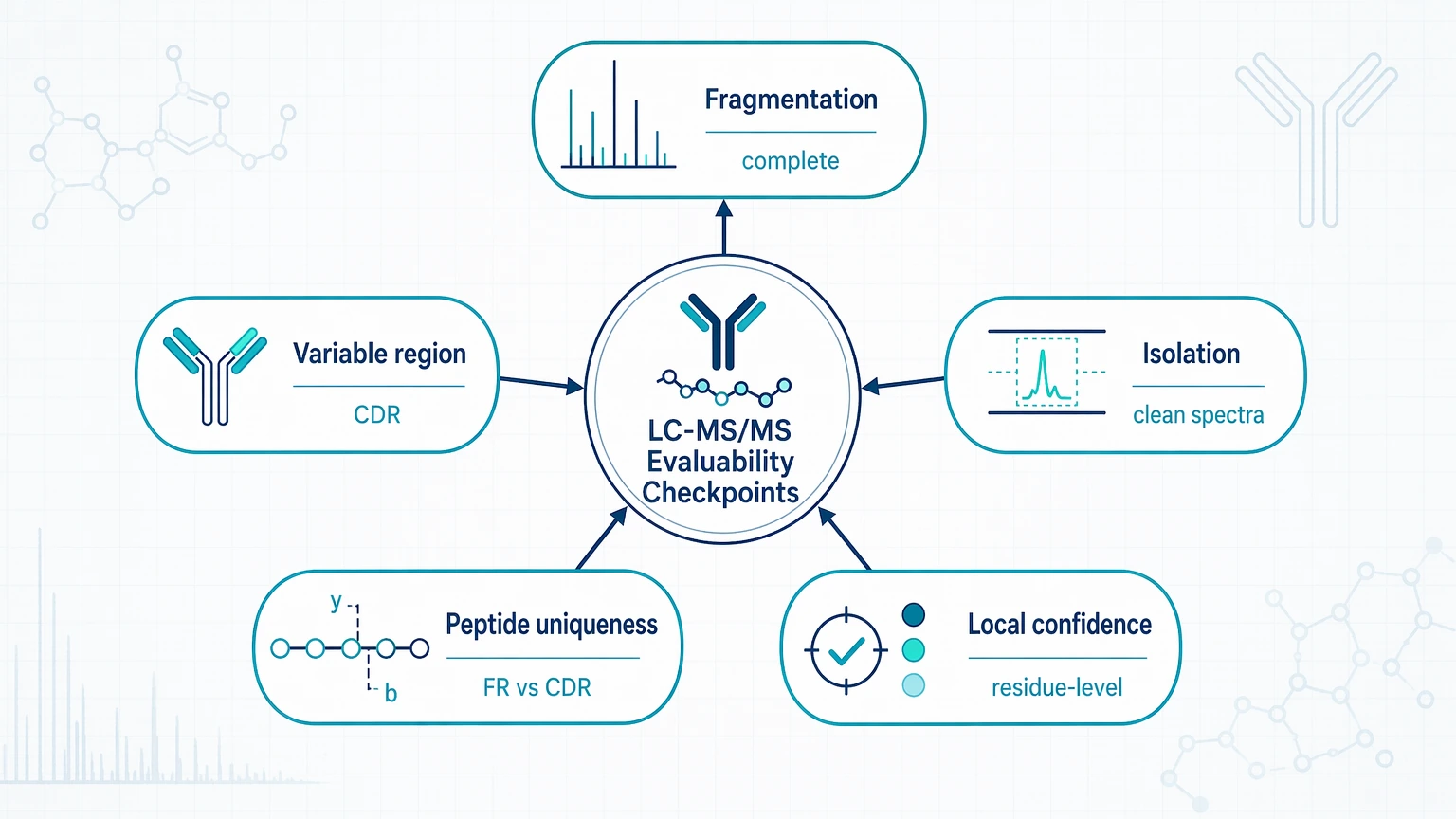
A compact decision table can help teams decide whether to continue with interpretation or correct the workflow first.
| Evidence check | What it supports | Main limitation | Next step |
|---|---|---|---|
| CDR peptide recovered with clean fragmentation | Residue-level review is justified | Local ambiguity may remain | Proceed to targeted de novo review |
| Good total coverage but missing variable-region peptides | Framework evidence only | Specificity question stays unresolved | Change digestion or acquisition plan |
| High unmatched spectra with contamination signals | Sample complexity is visible | False sequence paths become more likely | Improve cleanup or enrichment first |
| Mass shifts near known PTM-prone sites | Modification-aware interpretation is needed | Sequence variant and PTM can overlap | Map PTMs, then review unresolved spectra |
The takeaway is simple: an apparently complete dataset is not enough unless it contains interpretable evidence for the exact variable-region feature under review.
Step 4: Choose the right de novo scope
Not every case requires de novo protein sequencing. If the uncertainty is local, de novo peptide sequencing may be the better fit. If the project involves partly undocumented constructs, broader sequence gaps, or mixed evidence across multiple peptides, combining database search with de novo protein sequencing may be more informative.
The scope should match the question:
If your team has reached this decision point, MtoZ Biolabs can evaluate your project using the sample type, available LC-MS/MS data, unresolved region, and validation objective instead of forcing a generic sequencing plan.
Expected Results and Validation Methods
The most useful de novo output is not a simple statement that a sequence is solved. It is a structured interpretation package that separates immediate deliverables from follow-up confirmation.
Immediate deliverables
A good initial output may include candidate peptide sequences for unresolved regions, residue-level confidence maps with local drop zones, identification of Leu/Ile ambiguity or other isobaric residue limits, improved sequence coverage across the variable region, PTM-aware interpretation of spectra affected by glycosylation, deamidation, or oxidation, and comparison between database search assignments and de novo-supported alternatives. These deliverables can strengthen spectral interpretation and show whether a suspected sequence variant is plausible enough to affect antibody diversity or antibody specificity conclusions.
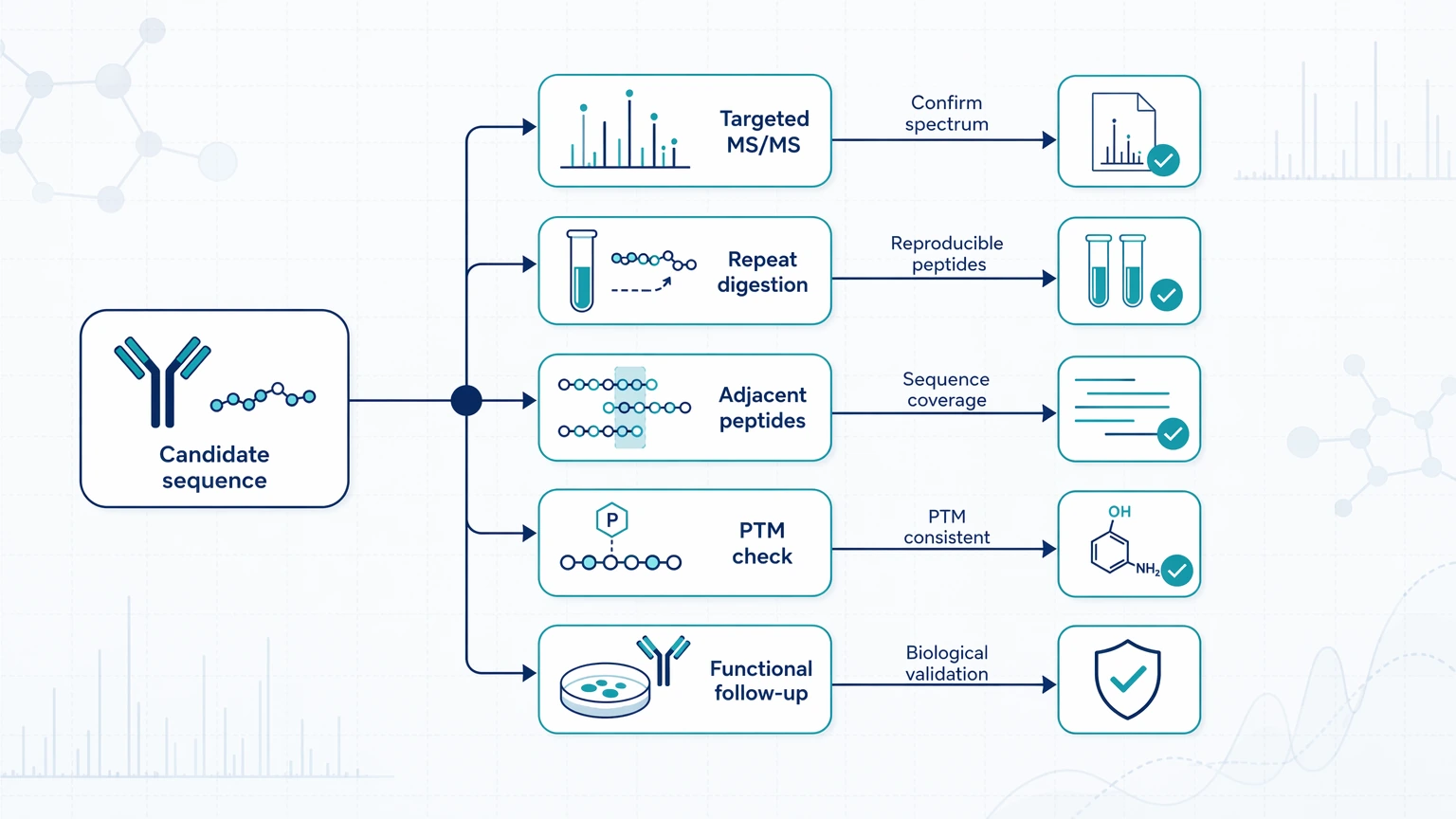
Follow-up confirmation
High-impact findings still need orthogonal validation. Suitable follow-up paths include targeted tandem mass spectrometry review of the candidate peptide, repeat digestion or complementary protease analysis to improve overlap, sequence consistency checks across adjacent peptides, orthogonal validation of PTMs when modification-driven ambiguity remains, and functional follow-up when the proposed sequence difference is meant to explain a specificity shift. Immediate de novo results can prioritize hypotheses and refine sequence interpretation, but they do not by themselves establish biological function or complete molecular identity.
Key Cautions and Practical Limits
De novo analysis is useful only within the limits of the material and data. Poorly purified, low-abundance, clipped, or aggregated samples may not yield the peptide evidence needed for confident variable region interpretation. Single-pass evidence is often insufficient for decisions tied to lead selection, impurity review, or specificity claims. Host-cell proteins, carryover, mixed antibody species, and preparation artifacts can distort unmatched-spectrum rates and generate false sequence paths. LC-MS/MS-based de novo workflows can also leave unresolved positions, especially with isobaric residues such as Leu and Ile. If the main question concerns intact mass heterogeneity, gross glycoform distribution, assay interference, or expression bias rather than local sequence content, intact protein mass spectrometry, targeted PTM mapping, or assay redesign may be more informative than de novo sequencing.
Common Mistakes That Delay Resolution
Three patterns slow interpretation repeatedly. First, teams often over-trust database search results in hypervariable regions; a matched peptide is not the same as strong support for a CDR call. Second, some ask de novo workflows to compensate for upstream weaknesses; if the sample is mixed or the digestion strategy misses CDR-containing peptides, more software will not repair the evidence gap. Third, teams stop after sequence inference; sequence evidence matters most when it leads to a next step such as orthogonal validation, clonotype reassessment, or reconciliation with binding data.
Service Routes to Consider
After the decision scope is clear, the next step is usually to route the project by evidence gap rather than by a broad service label.
FAQ
Can de novo sequencing help distinguish a true sequence variant from a PTM?
Often yes, but only when fragmentation supports that distinction. Deamidation, oxidation, and glycosylation can shift masses in ways that resemble sequence change. The interpretation is stronger when the modified state, neighboring fragments, and peptide overlap all support the same explanation.
Is broad sequence coverage enough to support antibody specificity conclusions?
Not by itself. What matters is whether the coverage includes the variable-region feature that could explain the observed binding behavior. Broad framework coverage with weak CDR evidence may still leave the specificity question unresolved.
When does clonotype information fail to explain the protein result?
This happens when the dominant expressed antibody species differ from the most abundant nucleic-acid-level signals, or when protein-level heterogeneity changes what is actually measured in binding or characterization assays. In those cases, protein evidence should be reviewed directly rather than inferred from repertoire composition alone.
Should teams repeat LC-MS/MS before requesting de novo interpretation?
If the current dataset has poor precursor isolation, low fragment intensity, or missing CDR-relevant peptides, repeating acquisition or changing digestion strategy is often the better first move. If the spectra are informative but database search remains weak, de novo interpretation is more justified.
Final Technical Takeaway
Antibody diversity and antibody specificity analysis become more stable when teams treat unresolved findings as a sequence-evidence problem at the right stage. De novo peptide sequencing or de novo protein sequencing is most suitable when ambiguity centers on a variable region feature, database search cannot explain the MS/MS evidence, and the answer will affect validation or engineering decisions. For teams comparing repertoire, binding, and LC-MS/MS outputs in a real antibody workflow, MtoZ Biolabs can submit your requirements into a project evaluation focused on sample condition, spectral evidence, unresolved sequence features, and the most appropriate confirmation path.
Related Services
Main Service |
Supporting Service |
Validation Service |
Alternative Service |
How to order?

