





How Does De Novo Sequencing Work? A B2B Guide to MS/MS Logic, Use Cases, and Escalation Points
- the peptide or protein region may be novel, engineered, species-uncertain, or absent from the reference database
- database search returns weak, conflicting, or biologically implausible matches
- the project needs database-independent identification rather than confirmation against a known sequence
- only partial sequence coverage is supported
- leucine/isoleucine ambiguity changes the project decision
- post-translational modification (PTM) burden disrupts the ion ladder
- the inferred sequence conflicts with intact mass or replicate spectra
- a ranked candidate sequence, annotated fragmentation spectrum, unresolved positions, and a validation plan if confidence is incomplete
- Ion-series continuity: consecutive fragment ions support residue order across part or all of the peptide.
- Mass accuracy: precursor and fragment measurements should fit the expected tolerance for the instrument and acquisition mode.
- Precursor isolation purity: the selected precursor should not be heavily mixed with co-isolated ions.
- Replicate consistency: repeated spectra should support the same sequence tag or candidate path.
- Modification-aware interpretation: neutral losses, diagnostic ions, and shifted fragment masses should fit a coherent PTM explanation.
- Unknown peptide identification: impurity peaks, natural isolates, or synthesis byproducts with no useful library match
- Engineered or edited sequences: therapeutic fragments, antibody-derived peptides, or designed constructs not represented in standard databases
- Species-uncertain samples: environmental, natural product, or nonmodel sources
- PTM-heavy analytes: modified peptides that no longer fit simple search assumptions
- Incomplete references: protein regions missing from the database or represented only partially
- one or more candidate sequences ranked by confidence score
- annotated fragmentation spectra
- sequence tags for high-confidence regions
- unresolved positions, including isobaric residue calls
- notes on precursor isolation purity, mass accuracy, and PTM interpretation
- repeat LC-MS/MS acquisition to test spectrum reproducibility
- complementary digestion to improve coverage or overlap
- intact mass comparison to check whether the inferred sequence fits the whole analyte
- targeted MS validation or synthetic peptide comparison for decision-critical residues
- terminal sequencing when peptide-level evidence does not resolve protein ends
- the spectrum supports only a partial sequence tag
- the inferred sequence conflicts with intact mass
- ambiguous residues remain at decision-critical positions
- peptide-level results do not answer a protein-level question
De novo sequencing is the right workflow when a team needs unknown peptide identification or sequence reconstruction from LC-MS/MS data and a database search cannot explain the spectrum with enough confidence. Instead of matching MS/MS data to a known entry, de novo sequencing reads the fragmentation spectrum directly, connects residue mass differences between fragment ions, and builds the most plausible amino acid path. When the spectrum shows a reasonably continuous b ion and y ion ladder, acceptable mass accuracy, and good precursor isolation purity, the output may support a high-confidence peptide call or at least a decision-useful sequence tag.
Quick Decision Block
Use de novo sequencing first when:
Escalate beyond de novo interpretation when:
Main output to expect:
What De Novo Sequencing Means in LC-MS/MS
In tandem mass spectrometry, a precursor ion is isolated and fragmented, and the resulting fragment ions are measured in an MS/MS spectrum. De novo sequencing interprets that fragmentation spectrum without relying entirely on a sequence database. The core question is simple: which amino acid order best explains the observed fragment pattern?
For de novo peptide sequencing, the most useful evidence often comes from b ion and y ion series. If two linked fragment ions differ by the mass of one residue, that residue mass difference becomes a candidate step in the sequence. When several steps line up across the spectrum, the analyst or software can assemble a sequence tag and extend it into a longer candidate peptide.
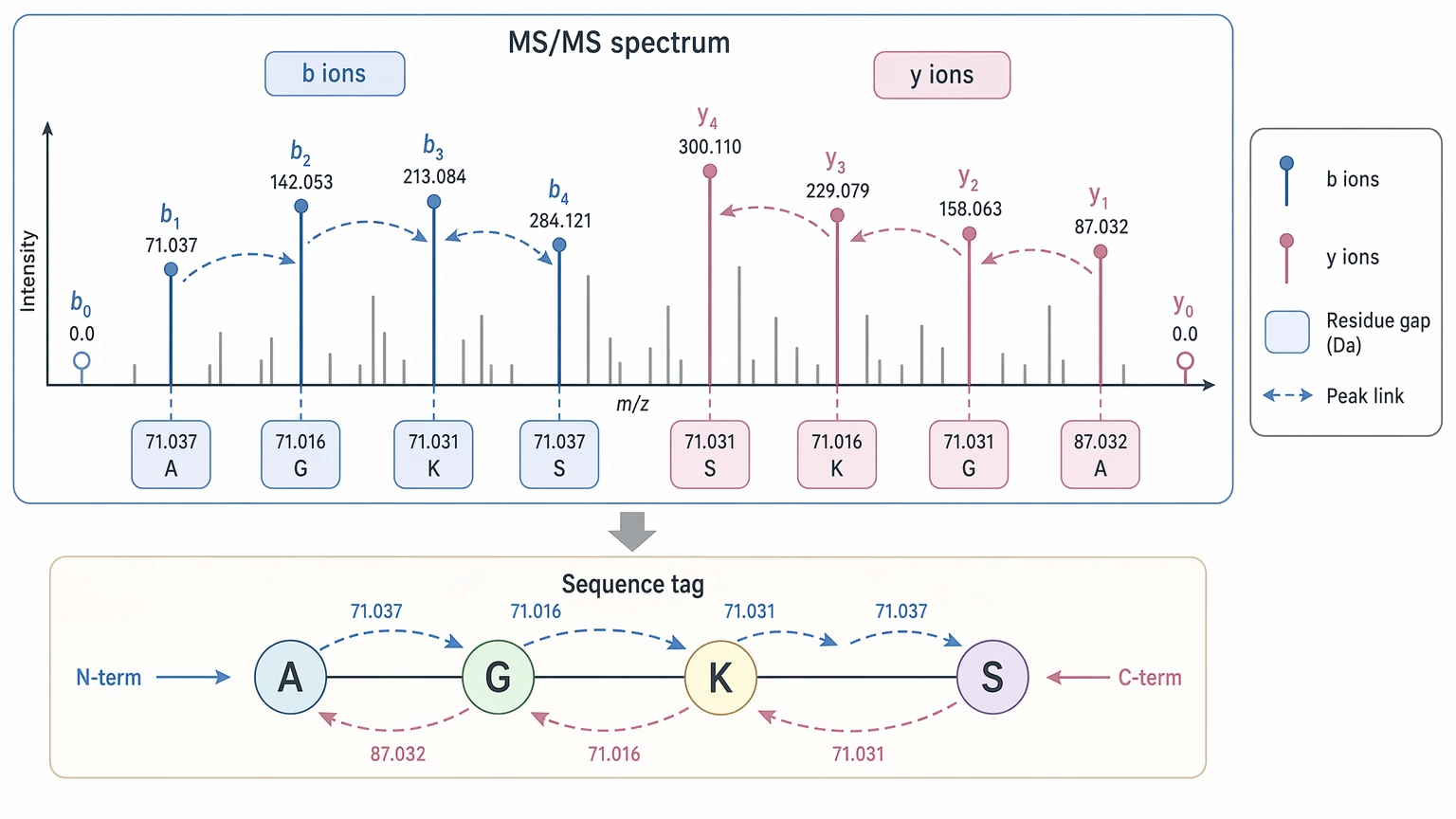
This is different from a database search, which asks which known peptide best fits the spectrum. De novo sequencing asks what sequence path fits the spectrum even if the peptide is not in the database. That distinction matters when the sample contains noncanonical sequences, variants, truncations, unexpected PTMs, or material from a poorly annotated source.
For de novo protein sequencing, the workflow is usually peptide-driven. Teams interpret multiple peptides first, then use overlap, complementary digestion, terminal information, and intact mass context to reconstruct larger protein regions. Routine bottom-up LC-MS/MS does not automatically deliver full de novo protein sequencing for every sample.
How MS/MS Logic Becomes a Sequence Call
The logic is straightforward on paper but demanding in practice.
First, the instrument measures the precursor ion mass. Next, fragmentation creates a set of fragment ions. The fragmentation spectrum is then checked for ion ladders and linked mass gaps that match amino acid residues. A continuous run of b ions, y ions, or both gives much stronger support than isolated peaks.
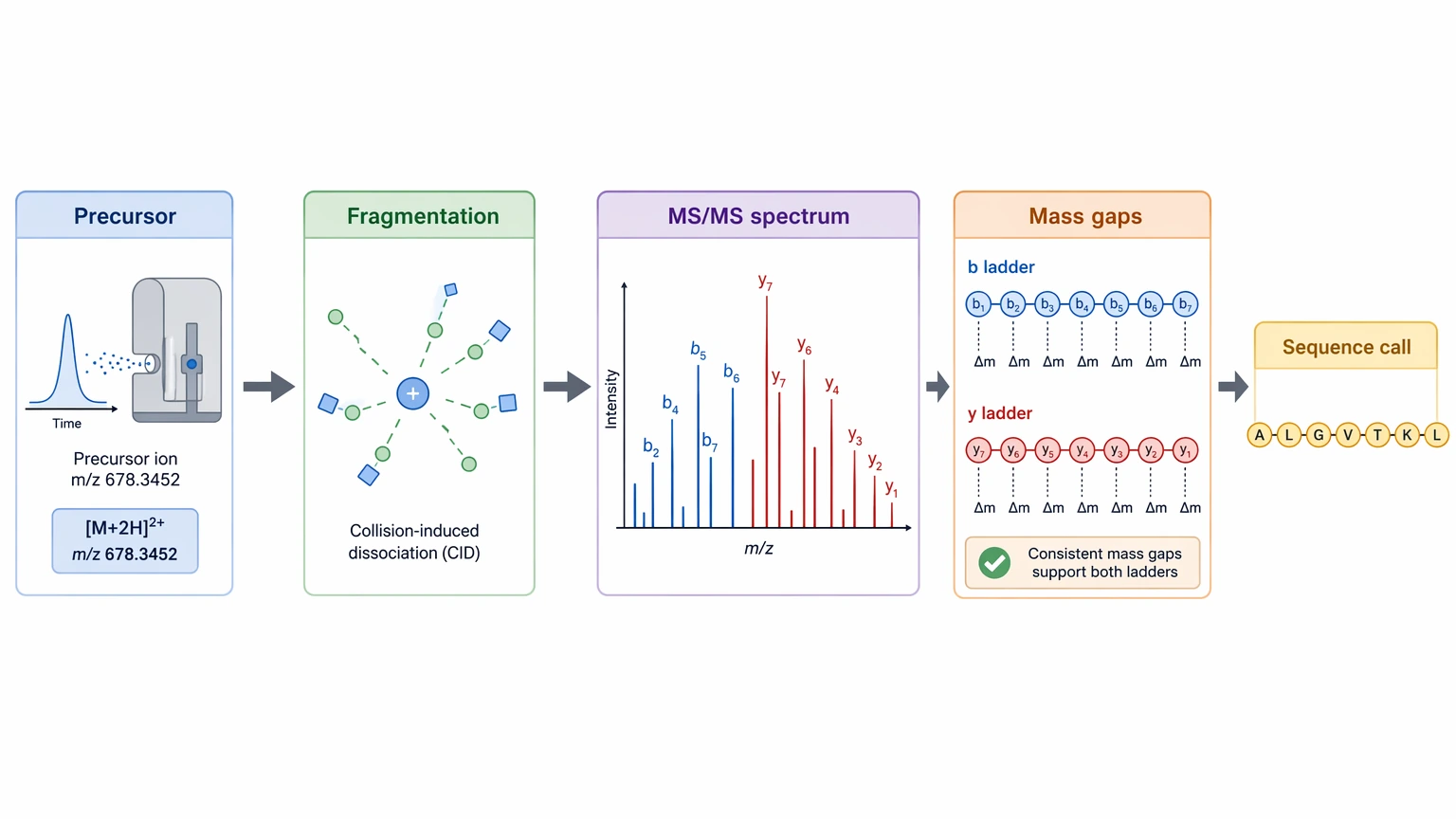
A credible de novo sequencing result usually rests on five evidence types:
One limitation needs to be stated plainly: standard MS/MS often cannot resolve leucine/isoleucine ambiguity, and PTM-rich spectra can support more than one residue path. In those cases, the confidence score may be local rather than full-length, and some positions may stay intentionally unresolved.
Why Database Search Can Fail
Database search remains efficient when the analyte is expected and the reference set is reliable. It becomes less decisive when the unknown itself is the main issue.
Common cases include:
For a project lead, the practical point is this: de novo sequencing is not “better” than database search across the board. It is more appropriate when reference matching no longer answers the real decision question.
When De Novo Sequencing Is a Good Fit
The table below helps teams choose the most suitable first interpretation path.
| Scenario | Recommended workflow | Main constraint | Next check |
|---|---|---|---|
| Unknown peptide with failed database search | Start with de novo peptide sequencing | Missing ion ladder can limit full assignment | Repeat LC-MS/MS if confidence is low |
| Engineered peptide or variant region | Combine de novo sequencing with intact mass review | PTMs may compete with designed edits | Confirm critical residues |
| Protein with incomplete sequence information | Peptide-level de novo plus complementary digestion | Full reconstruction may remain partial | Add terminal or overlap evidence |
| PTM-rich analyte | Modification-aware de novo interpretation | Localization may remain uncertain | Use orthogonal validation |
| Standard sample with trusted reference database | Use database search first | De novo may add effort without added value | Reserve for unresolved spectra |
The takeaway is practical: start with de novo sequencing when sequence novelty is the main uncertainty, not when a standard reference workflow already answers the question.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
What a Technical Team Should Check Before Approval
Before approving a project, look at the evidence quality, not just the method label.
| Evidence | What it supports | Common limit | Practical response |
|---|---|---|---|
| Continuous b ion / y ion ladder | Residue order | Gaps weaken terminal assignment | Extend with replicates |
| Good precursor isolation purity | Cleaner precursor-fragment relationship | Chimeric spectra can create false paths | Reacquire with tighter isolation |
| Consistent replicate spectra | Confidence assignment | Inconsistency suggests unstable interpretation | Review sample purity and acquisition |
| Intact mass agreement | Sequence plausibility | PTMs or truncations may still complicate fit | Add modification-aware review |
| Diagnostic PTM ions | Modified residue interpretation | Site assignment may remain partial | Confirm with targeted follow-up |
If your group already has LC-MS/MS files and needs to decide whether they can support reportable sequence inference, MtoZ Biolabs can evaluate your project and help you submit your requirements around sample amount, purity, fragmentation evidence, and expected deliverables before full de novo sequencing work begins.
Expected Results and How to Validate Them
A useful de novo sequencing report should separate immediate deliverables from follow-up confirmation.
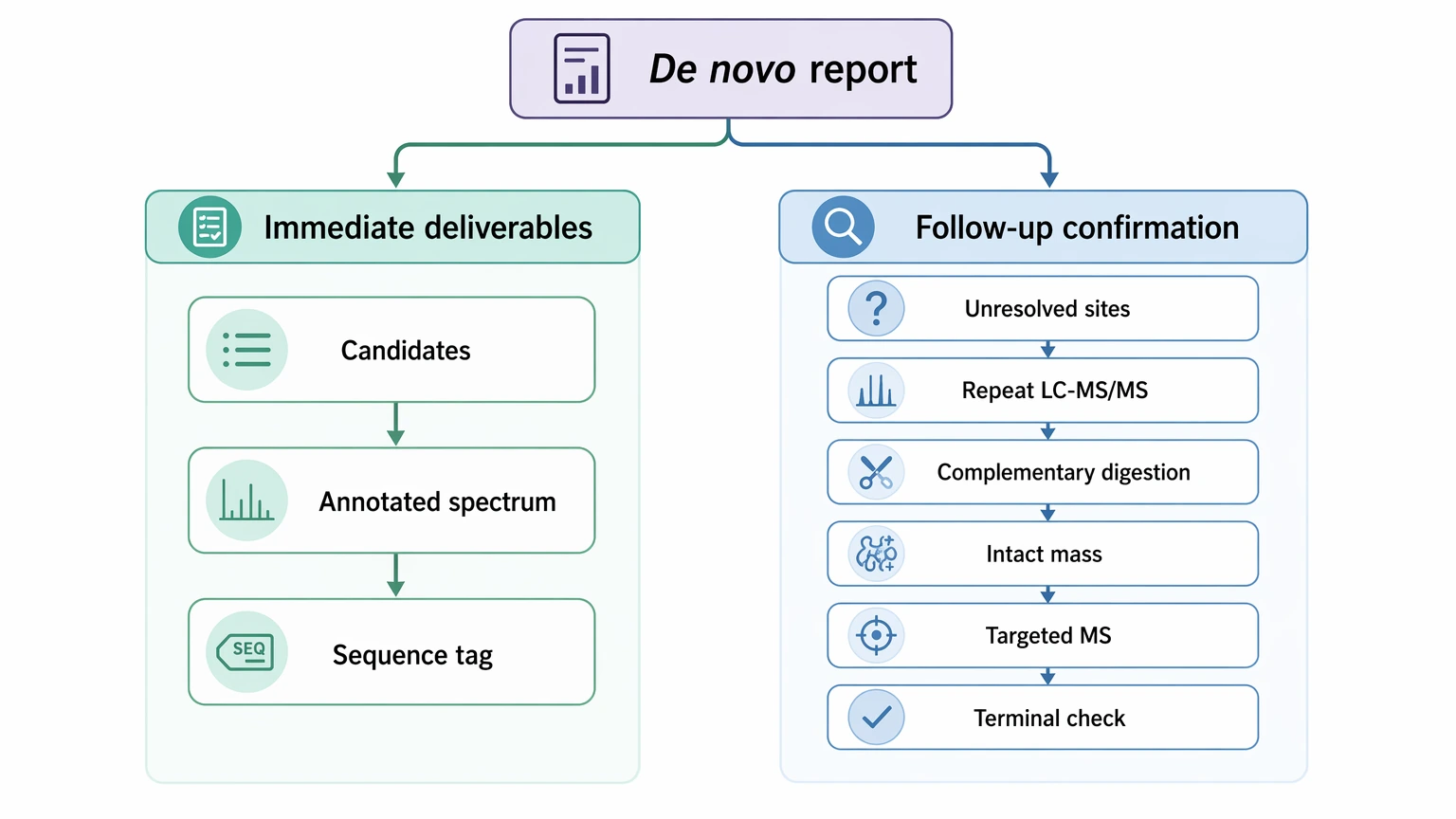
Immediate deliverables may include:
Follow-up confirmation may include:
A short sequence tag can still be actionable if the project only needs family-level assignment, impurity narrowing, or a decision on whether deeper sequencing is worth funding. That is different from a full peptide call, and the confidence assignment should always be read in that context.
Key Cautions and Practical Limits
De novo sequencing is useful precisely because it works without full database dependence, but it still has clear boundaries.
Sample quality or amount limits: low abundance, poor enrichment, degradation, or contaminated preparations reduce the chance of getting interpretable fragmentation spectra.
Controls and repeat expectations: a single attractive spectrum may not be enough for a business-critical decision. Replicate consistency usually matters more than one visually clean spectrum.
Batch or contamination risk: low precursor isolation purity can produce chimeric spectra, especially in complex mixtures. False residue paths can come from co-isolated ions rather than the target precursor.
Interpretation boundaries: longer peptides, incomplete fragmentation, and unknown PTMs can reduce sequence confidence. De novo sequencing can return partial sequence coverage even when the spectrum is informative.
When another method is the better next step: if the main need is routine reference-matched identification, use a database search first. If the unresolved issue is terminus definition, intact proteoform structure, or a residue call that standard MS/MS cannot discriminate, terminal sequencing, top-down design, or targeted orthogonal validation may be the better next step.
Escalation Points That Matter in Real Projects
Escalation is not a failure state. It is the point where the inferred sequence alone no longer supports the project decision.
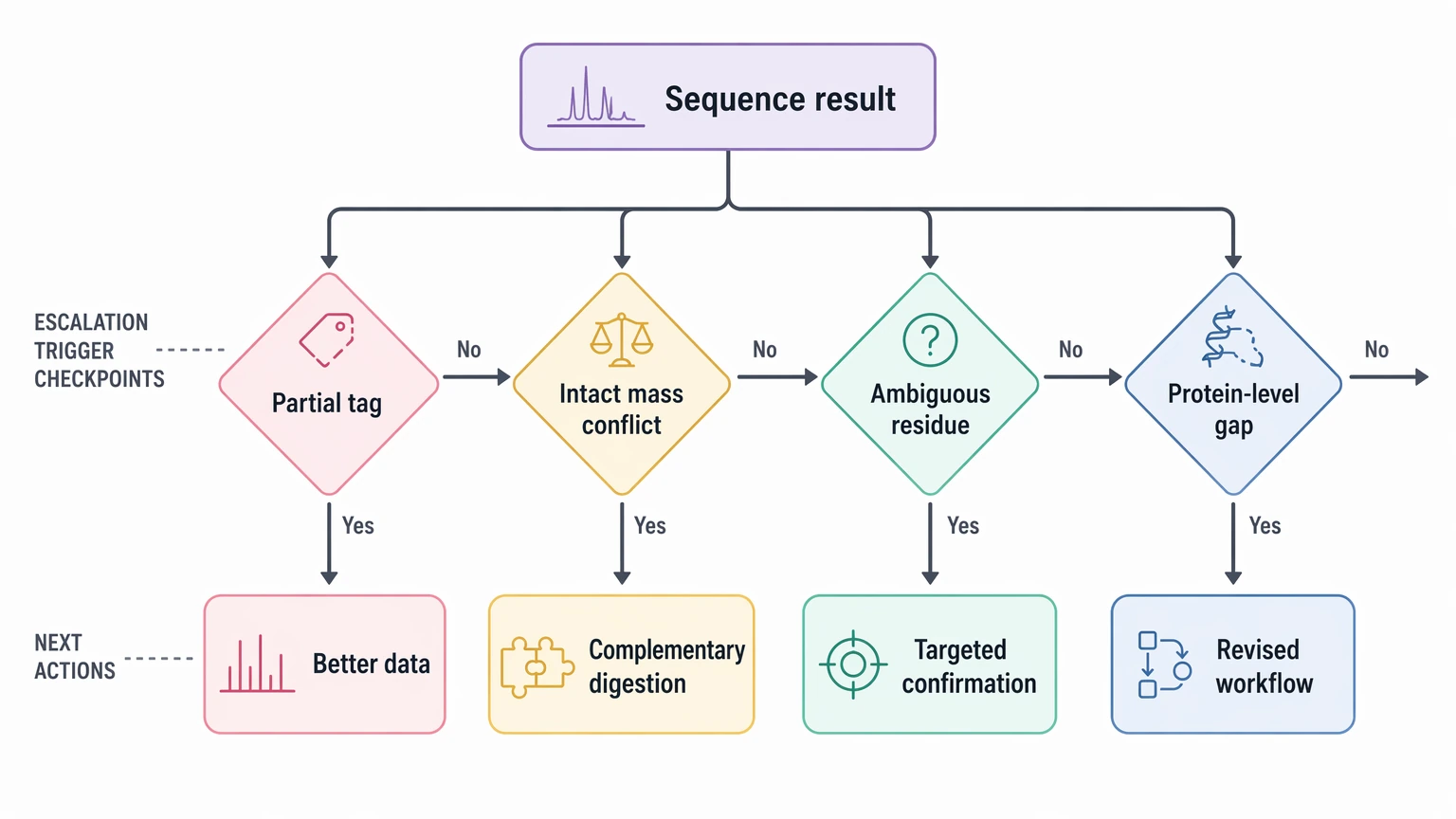
The most common triggers are:
At that stage, the next move is usually better data, complementary digestion, targeted confirmation, or a revised workflow rather than more informal interpretation. If you need a staged plan that compares sequence-tag reporting with validation-focused follow-up, contact MtoZ Biolabs to evaluate your project and discuss whether de novo peptide sequencing, de novo protein sequencing, targeted confirmation, or an alternative route fits the sample and decision goal more closely.
Conclusion
De novo sequencing works by translating a fragmentation spectrum into a residue-by-residue sequence hypothesis through b ion and y ion relationships, residue mass difference logic, mass accuracy context, and spectrum annotation. It is most suitable when the team faces unknown peptide identification, nonreference sequences, engineered variants, or database-search failure. Its limits matter more when fragmentation is incomplete, precursor isolation purity is poor, PTMs create competing interpretations, or the project depends on resolving residues that standard MS/MS may leave ambiguous. For peptide and protein characterization projects, the most useful planning sequence is to define the required deliverable, compare that goal with the actual LC-MS/MS evidence, and then choose between direct reporting, additional acquisition, orthogonal validation, or another sequencing route. If your team is preparing an unknown-sequence project, bring sample type, amount, purity, intact mass expectations, and existing LC-MS/MS files into the consultation so the next step matches the real decision at hand.
FAQ
When is a partial sequence tag enough to move a project forward?
A partial tag is often enough when the goal is to narrow an impurity, distinguish a nonreference peptide from a known product, or justify a targeted follow-up experiment. It is less sufficient when release testing, IP-sensitive residue positions, or exact terminal structure matter.
Can de novo sequencing distinguish a novel sequence from an unexpected PTM?
Sometimes, but not always. A novel residue path and a modified known peptide can support overlapping explanations. Intact mass checks, replicate spectra, and PTM-aware interpretation are often needed before choosing between those two possibilities.
Is de novo protein sequencing the same as analyzing one peptide spectrum?
No. De novo protein sequencing usually combines multiple peptide-level results, overlap logic, and added contextual evidence. One peptide spectrum may identify a region, but it rarely defines an entire protein sequence on its own.
What should a buyer ask before sending samples or raw data?
Ask what sample amount is likely needed, whether the material is purified or mixed, whether existing LC-MS/MS data meet precursor isolation purity and fragmentation requirements, how unresolved positions will be reported, and what confirmation steps are available if the first pass returns only partial sequence coverage.
How should teams read a confidence score in a de novo report?
Treat it as evidence strength for a specific sequence path, not as a blanket guarantee. Local confidence may be high for a sequence tag while terminal assignment or PTM localization remains uncertain.
How to order?

