





De Novo Sequencing vs Resequencing: Which Strategy Is Better for Novel Sequence Discovery Projects?
- Start with de novo sequencing when no reliable reference sequence exists or when real novelty is expected.
- Start with resequencing when a likely parent sequence exists and the decision centers on limited changes.
- Use a hybrid workflow when you have a partial reference, likely post-translational modification (PTM) burden, uncertain termini, or a mixed proteoform population.
- Escalate validation early when the downstream use includes synthesis, impurity attribution, construct redesign, or targeted follow-up.
- Treat every output as evidence-bounded: an LC-MS/MS-derived candidate sequence may still contain ambiguous residue assignment, especially around termini, PTMs, or sparse fragmentation.
- one or more ranked candidate sequence hypotheses
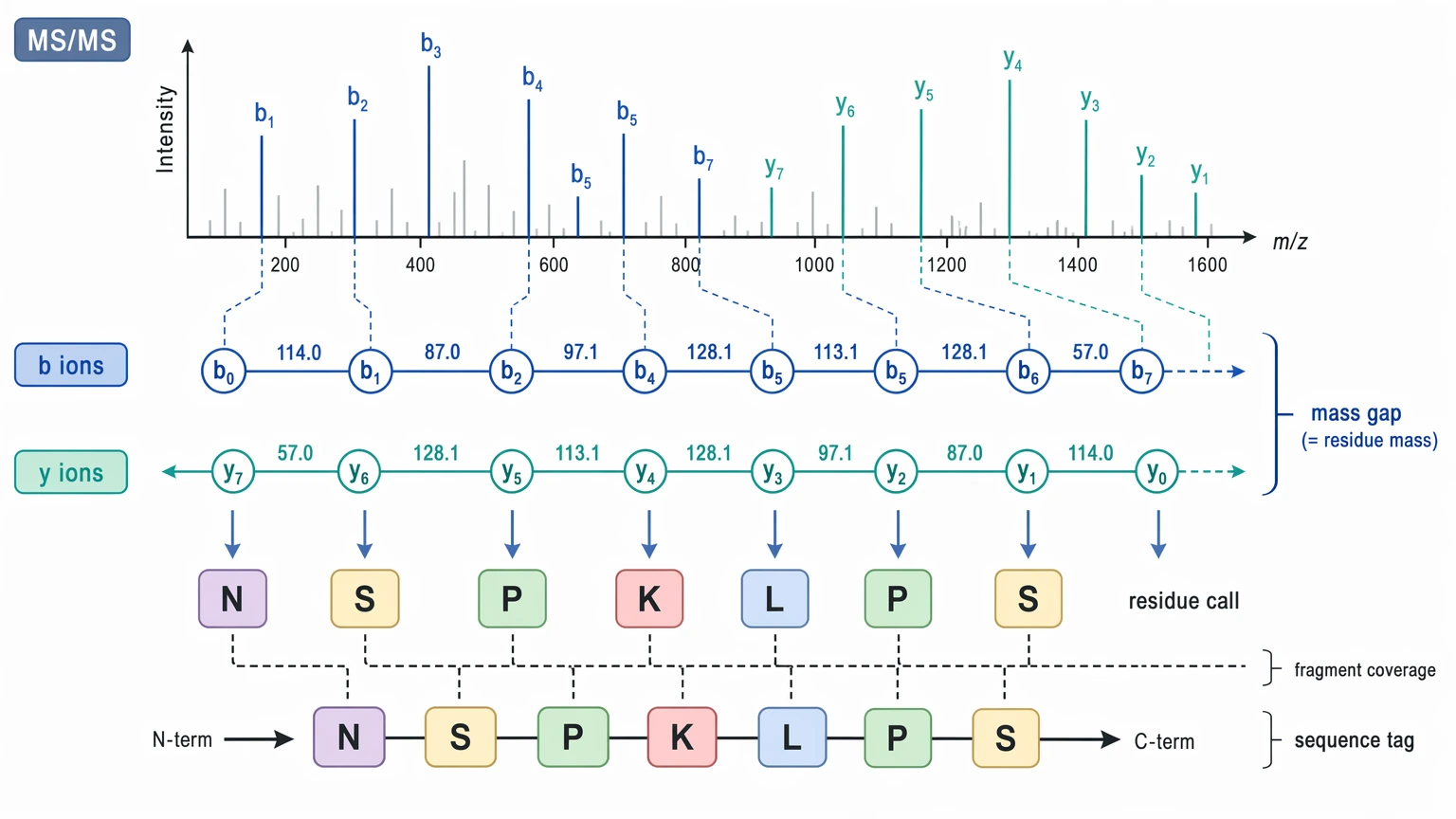
- residue-supported maps linked to b ions / y ions
- notation of ambiguous residue assignment
- explicit marking of leucine/isoleucine ambiguity
- PTM-aware alternatives where the mass shift is not uniquely attributable
- confidence comments tied to local fragment-ion coverage
- recommendations for the next confirmation step
- targeted MS confirmation of a proposed variant region
- synthesis matching for a short peptide candidate
- N- or C-terminal confirmation when termini drive the decision
- top-down or intact-mass support when proteoform structure is still in question
- mutation-informed or construct-informed follow-up when a parent design is partly known
Choose de novo sequencing first when your LC-MS/MS data needs to recover a peptide or protein sequence that may include non-reference regions, unexpected truncation, or modification patterns that a database search cannot account for. Choose resequencing first when you already have a credible reference sequence and the main question is whether the analyte differs by a limited number of substitutions, terminal changes, or local edits. If your sample sits somewhere between those two cases, a hybrid workflow is often the safer place to start: constrain what you can with resequencing, then apply de novo sequencing only where the evidence still does not settle the call.
That decision matters because novel sequence discovery is not just about generating more sequence candidates. It is about managing sequence confidence, limiting reference bias, and deciding how much orthogonal validation you need before anyone treats a proposed sequence as actionable.
Quick decision block
Use this compact guide near project kickoff:
What the two strategies mean in LC-MS/MS projects
In peptide sequencing and protein sequencing by tandem mass spectrometry, de novo sequencing infers residue order directly from the MS/MS spectrum. The method relies on interpretable ion series, especially b ions / y ions, local mass differences, and enough fragment-ion coverage to support residue order without needing a full database match.
Resequencing uses a known or suspected parent sequence as the frame for interpretation. In practice, it asks whether the analyte is close to that reference and, if so, where the differences are located. That framing can speed up analysis, but it also creates a real risk: if the true analyte includes sequence content outside the assumed model, the workflow may pull the result toward a familiar answer instead of a complete one.
For novel sequence discovery, the main distinction is not some abstract split between “unbiased” and “biased.” The real question is whether the evidence on hand and the project goal support direct sequence inference or a reference-guided approach.
Comparison by the decision dimensions that matter most
Reference availability
This is the first and most important divide. If the only sequence information available is a distant homologous sequence, resequencing can narrow the search space, but it can also overfit the data. De novo sequencing becomes more attractive as confidence in the reference drops.
Expected novelty
If your project may contain novel processing products, non-reference segments, or unexpected sequence drift, de novo sequencing has the stronger discovery role. Resequencing is more efficient when novelty is likely to stay local, such as one or a few substitutions, a cleavage event, or terminal trimming.
Spectral quality and sample limits
Both strategies benefit when the MS/MS spectrum shows continuous ion ladders across the sequence. De novo sequencing depends on that continuity more heavily. When sample amount is limited, precursor isolation is imperfect, or reruns are constrained, resequencing often gives a cleaner first pass if a trusted reference is already available.
PTM burden
PTMs complicate both strategies. A known PTM target can be built into interpretation, but unexpected PTMs can look like substitutions or processing events. One practical limit should stay explicit here: MS/MS interpretation may not fully separate sequence variation from PTM-related mass shifts in every region, especially when fragment coverage is incomplete.
Validation burden
If the output will guide synthesis, functional testing, impurity root-cause work, or regulatory-facing characterization, the better workflow is the one that leaves fewer high-impact unknowns. That does not automatically mean resequencing. It means choosing the route that gives you the most defensible candidate sequence for the question you actually need answered.
Scenario comparison for project selection
Use the table below to choose a starting workflow rather than a final answer.
| Scenario | Better-fit strategy | Why | Main limitation | Validation follow-up |
|---|---|---|---|---|
| No usable reference; likely unknown peptide | De novo sequencing | Directly addresses non-reference content | More unresolved positions if fragmentation is patchy | Targeted MS confirmation or synthesis match |
| Known parent sequence with a few expected changes | Resequencing | Efficient for localizing limited differences | May miss unexpected non-reference regions | Site-focused confirmation |
| Partial reference with uncertain N- or C-termini | Hybrid workflow | Uses reference where helpful but keeps open discovery at ends | More interpretation branches | Terminal sequencing support or region-specific confirmation |
| PTM-rich analyte with substitution-like mass shifts | Hybrid workflow | Balances PTM-aware interpretation with local de novo calls | PTM localization may stay uncertain | PTM-targeted follow-up and replicate review |
| Mixed proteoforms or low-purity isolate | Cautious resequencing if parent is known; otherwise focused de novo sequencing | Limits overcalling from mixed spectra | Co-eluting species reduce confidence for both routes | Cleanup, fractionation, or orthogonal fraction confirmation |
Takeaway: the best first choice is the workflow that removes the most important uncertainty in your specific discovery scenario.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
Where false confidence usually enters the report
Most project errors do not come from choosing the “wrong” label. They come from reading beyond what the evidence can support.
In resequencing, false confidence often begins with a plausible but incorrect reference sequence. A near match can make the residue map look cleaner than the analyte really is. Unexpected inserts, heavy processing, or non-reference terminal regions may then be undercalled because the model already assumes what the answer should look like.
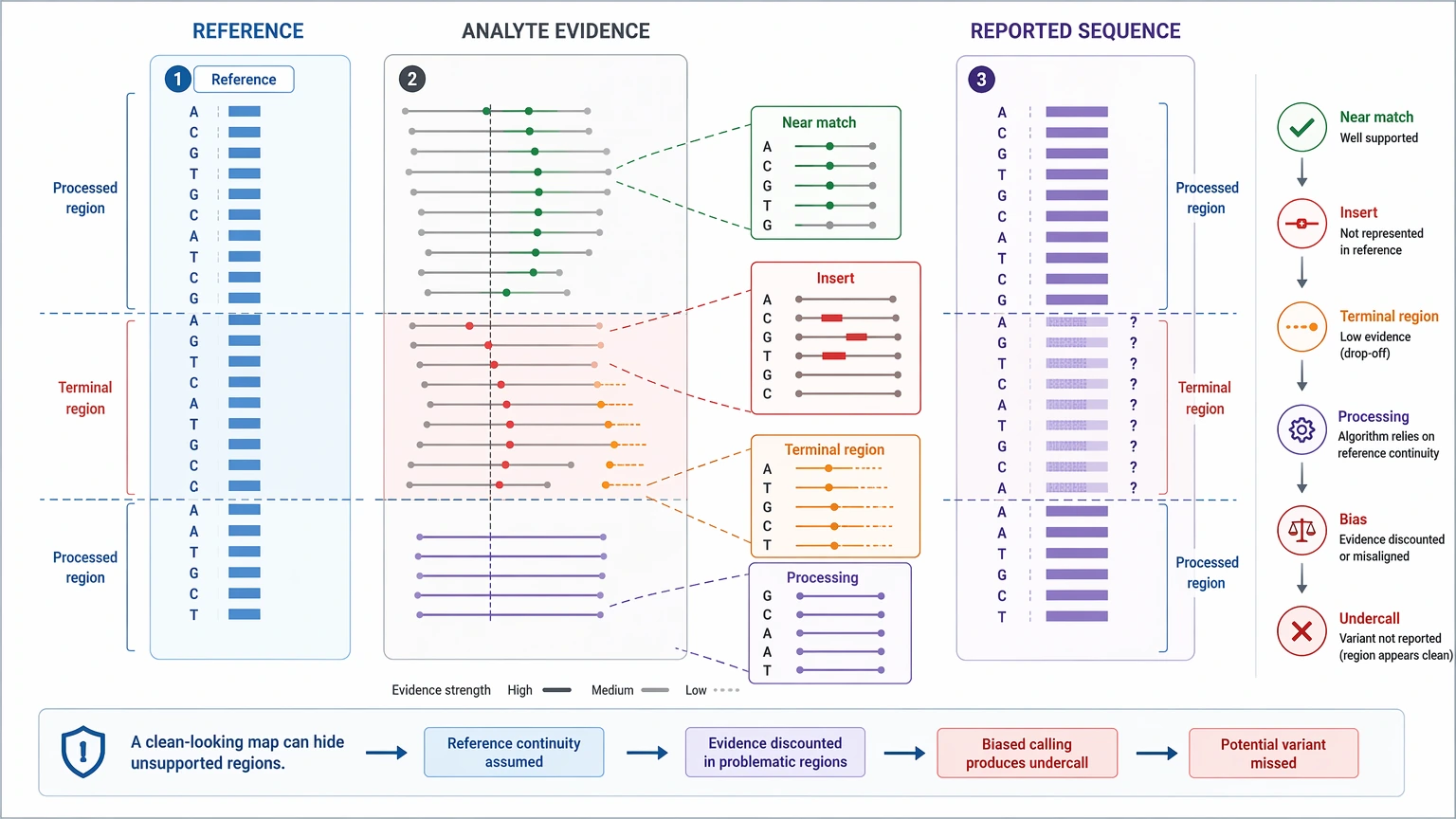
In de novo sequencing, false confidence usually shows up when a strong sequence tag gets treated like a full-length solution. One well-supported segment does not guarantee that the rest of the molecule is correct. Confidence often falls at the N-terminus, C-terminus, across low-intensity regions, or around modifications that create competing interpretations.
Another hard limit should stay visible in any discovery report: standard LC-MS/MS in bottom-up proteomics often cannot resolve leucine/isoleucine ambiguity by mass alone. That is not a reporting flaw. It is a real limit of the method unless additional evidence is introduced.
Expected results and validation methods
A useful discovery report should separate immediate deliverables from follow-up confirmation.
Immediate deliverables
At the end of the primary workflow, expect outputs such as:
These deliverables are decision tools. They show what is well supported, what is still tentative, and which region deserves the next experiment.
Follow-up confirmation
Follow-up confirmation should answer a narrower question than the initial discovery run. Suitable options may include:
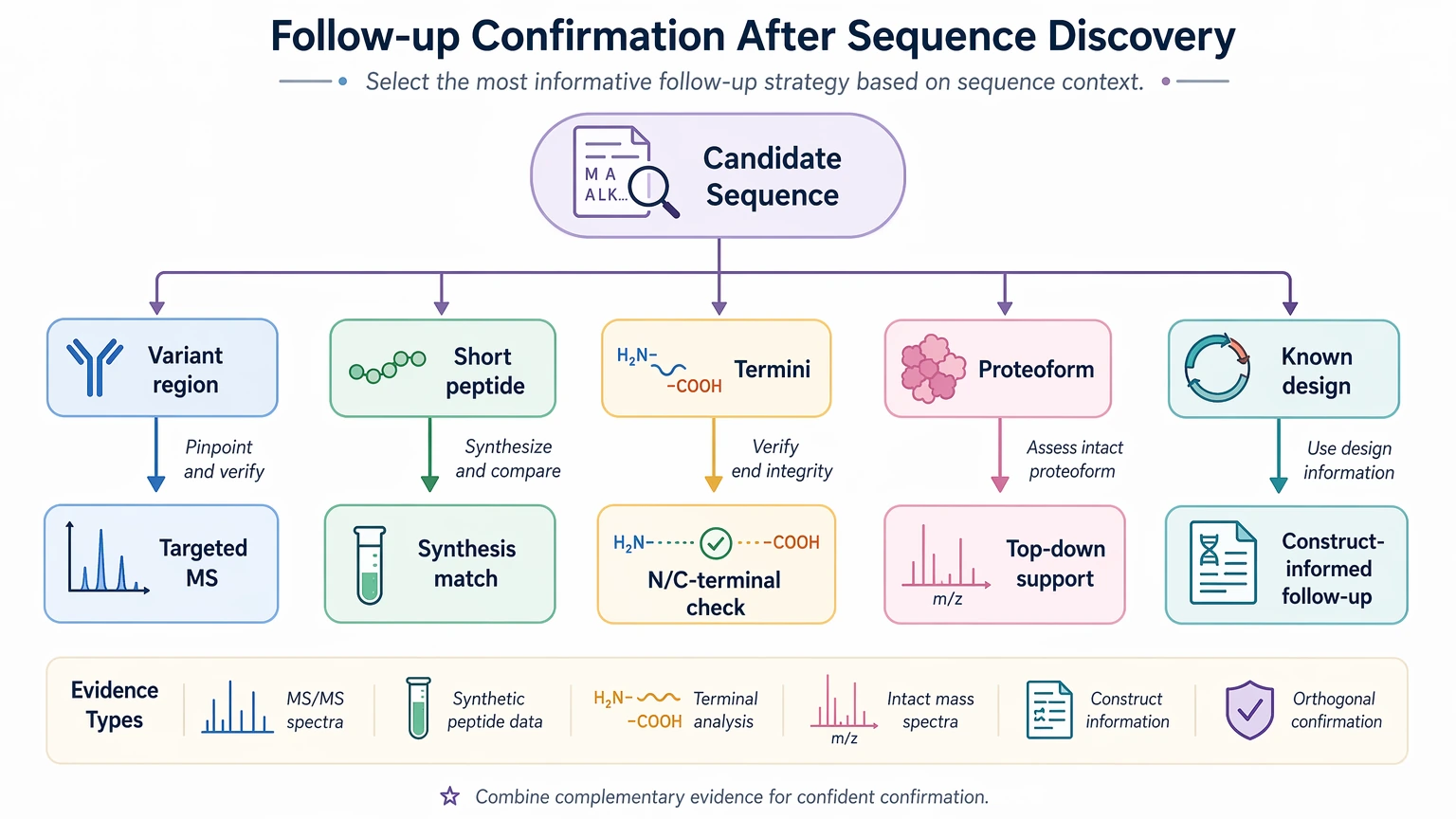
If your team needs to decide between a direct discovery workflow and a reference-guided one before committing scarce sample, you can submit your requirements to MtoZ Biolabs for evaluation against existing LC-MS/MS data, expected sequence novelty, and the depth of confirmation your project will need.
Key cautions and practical limits
Several constraints should shape the plan before instrument time is spent.
Sample quality or amount limits
Low purity, mixed species, and very limited material reduce sequence confidence for both strategies. De novo sequencing is usually affected more because weak or composite spectra break residue continuity faster.
Controls and repeat expectations
Replicate spectra, alternate charge states, and digestion or fractionation controls can change confidence substantially. Without them, a plausible sequence path may remain only a hypothesis.
Batch and contamination risk
Carryover, co-isolated precursors, and contaminated fractions can create misleading fragment patterns. This matters most when the project is trying to claim novelty from a low-abundance feature.
Interpretation boundaries
Database-independent does not mean ambiguity-free, and reference-guided does not mean correct by default. PTMs, long peptides, weak terminal fragments, and mixed proteoforms can all leave more than one interpretation on the table.
When another method is the better next step
If the decision depends on residue-level certainty at a poorly fragmented terminus, complete PTM localization, or separation of mixed proteoforms, another method may be the better next step. In some projects, targeted confirmation, terminal sequencing, improved purification, or outside workflow review will do more than another broad discovery pass.
A practical way to choose the starting workflow
Start with the question the sequence must answer, not with the method that sounds most sophisticated.
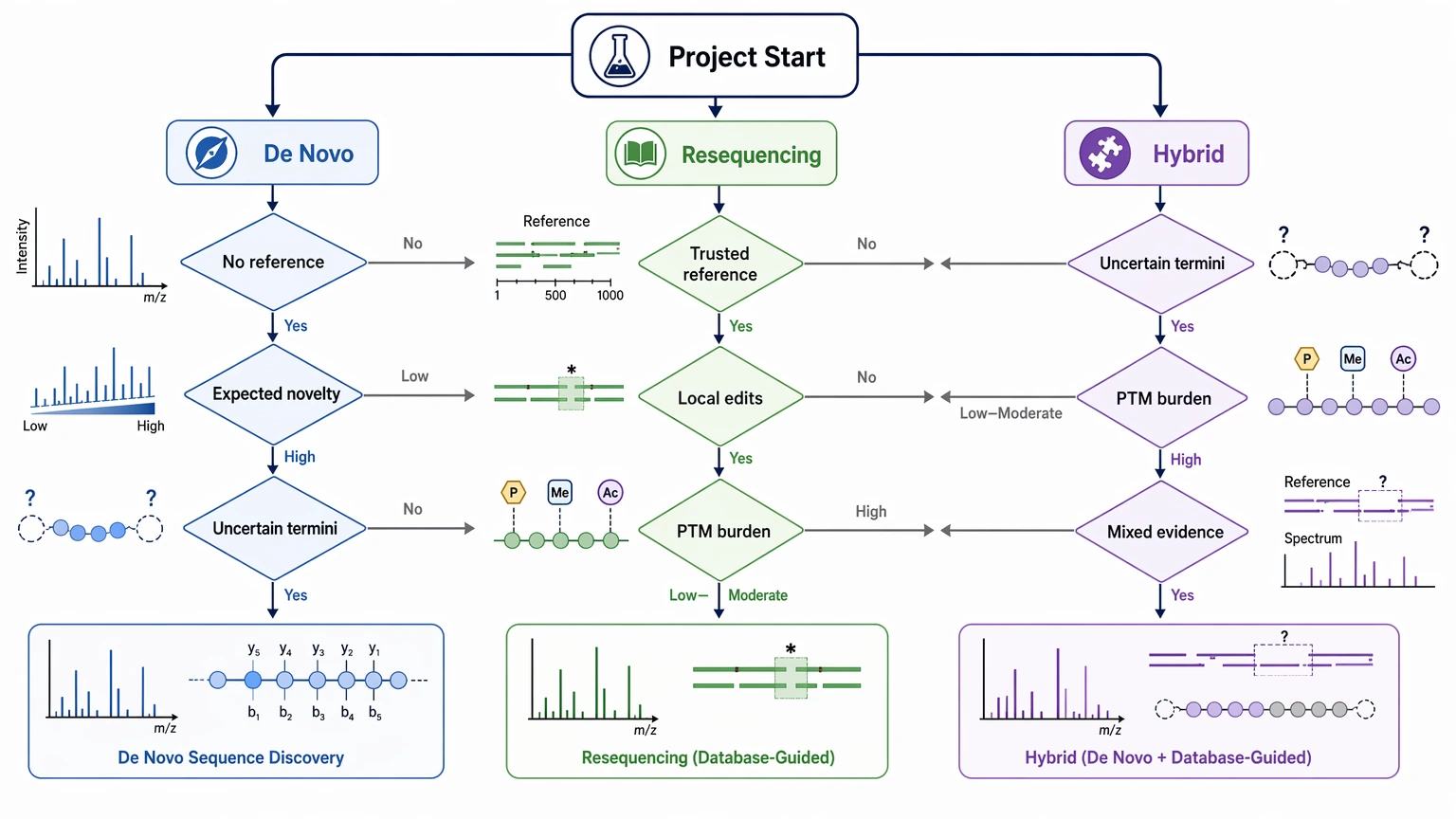
Use de novo sequencing when the project cannot lean on a reference and novelty itself is the main target. Use resequencing when the sequence question is narrower and the parent model is credible. Use a hybrid workflow when the evidence is partly reference-supported but the highest-risk regions still need open interpretation.
In operational terms, the right workflow is the one that turns LC-MS/MS evidence into a report your downstream team can act on without overstating what is known. For impurity studies, natural peptide isolation, degraded therapeutics, recombinant drift checks, and PTM-rich samples, that usually means choosing a strategy that keeps uncertainty visible instead of smoothing it over.
Technically, de novo sequencing is best suited to discovery-heavy questions, resequencing is best suited to reference-near variation, and a hybrid path is often best when the sample falls in between. If your project involves limited material, incomplete references, or a short validation window, contact MtoZ Biolabs to evaluate your project and discuss which workflow, deliverable format, and confirmation plan fit the evidence you actually have.
FAQ
How much reference information is enough to make resequencing worthwhile?
A useful reference does not need to be perfect, but it should be biologically plausible and close enough to the expected analyte that local differences can be interpreted without forcing the whole sequence into the wrong model.
Does a failed database search automatically mean de novo sequencing is required?
No. A weak database search can also reflect PTM complexity, poor precursor isolation, digestion problems, or mixed species. Resequencing may still be the better first move if a strong parent sequence is available.
Can a hybrid workflow save sample?
Often yes. If reference-guided analysis can narrow the candidate space first, you may be able to reserve limited reruns for the ambiguous regions instead of spending material on broad reanalysis.
What makes a candidate sequence actionable enough for synthesis?
Usually a short list of conditions: the high-value region is well supported by fragment ions, major alternative interpretations have been pushed down, and the remaining ambiguity does not affect the synthesis or test objective.
When should project teams worry most about leucine/isoleucine ambiguity?
It matters most when the exact residue identity changes biological interpretation, construct design, IP decisions, or a synthesis plan. If that distinction is not critical to the decision, the ambiguity can sometimes be documented and managed rather than eliminated right away.
Is top-down support always needed after de novo sequencing?
No. It is most useful when intact proteoform context, terminal structure, or modification pattern is central to the decision and bottom-up evidence leaves too many competing explanations.
How to order?

