





De Novo Sequencing vs Database Search: Which Workflow Fits Novel Peptides, PTMs, and Low-Reference Discovery Projects?
- The sample has a biologically close and reasonably complete reference.
- The main question is peptide identification rather than novelty calling.
- The PTM scope is limited enough to search without opening an unmanageable space.
- Absent database entries: the true peptide is missing from the searchable sequence set.
- Over-expanded PTM settings: too many modification possibilities blur ranking and inflate the search space.
- Forced assignments of unexpected products: a real novel cleavage product or engineered variant gets mapped to the nearest known peptide instead of the correct one.
- Run database search under biologically sensible settings.
- Isolate unexplained, weakly assigned, or high-interest spectra.
- Derive sequence tags or de novo candidates from the best spectra.
- Build or refine a custom database and re-search with tighter hypotheses.
- Plan confirmation around the strongest candidates.
- the sequence is novel,
- the modification pattern is novel, or
- the spectrum is not informative enough to support either claim.
- Sample quality or amount limits: low-abundance peptides, mixed backgrounds, or insufficient material can reduce fragment-ion continuity and weaken de novo inference.
- Controls and repeat expectations: novelty claims are stronger when the same candidate appears across technical or biological repeats, or when controls help rule out background peptides.
- Batch and contamination risk: carryover, digestion artifacts, and mixed-source contamination can produce misleading spectra that look novel at first glance.
- Interpretation boundaries: leucine/isoleucine ambiguity, incomplete ion ladders, and uncertain PTM localization should be disclosed rather than hidden.
- When another method is better: top-down approaches, transcript-supported proteogenomics, or targeted follow-up may be the better next step when bottom-up MS/MS alone cannot support the claim you need.
Start with database search when your LC-MS/MS dataset maps to a credible reference proteome and the main deliverable is confident peptide identification for expected sequences. Shift toward de novo sequencing when the real peptide may not exist in the reference, when sequence novelty is part of the study goal, or when standard search settings no longer explain PTM-rich or processing-derived spectra. In many discovery projects, the most defensible option is a hybrid workflow that combines database search, sequence tag generation, focused re-searching, and planned orthogonal validation.
Quick workflow decision block
| Project signal | Best first choice | Why | What to watch |
|---|---|---|---|
| Well-annotated species, expected peptides, narrow PTM scope | Database search | Strong peptide-spectrum match (PSM) framework and established false discovery rate (FDR) control | Missed peptides if the true sequence is absent |
| Low-reference organism or divergent sequence content | De novo sequencing or hybrid | Less dependence on database completeness | Higher sequence ambiguity and validation burden |
| Known sequence backbone, but PTM-focused question | Database search with constrained variable modification settings | Efficient for expected modifications | Search space expands quickly if PTM settings are too broad |
| Unexpected mass shifts or novel processing products | Hybrid | Lets you test both reference-supported and inference-driven explanations | PTM identity and PTM localization may stay provisional |
| Novel peptide discovery is the main output | Hybrid leaning toward de novo sequencing | Better fit for absent-reference candidates | Follow-up confirmation is usually needed before strong biological claims |
This comparison matters most when a dataset falls between routine identification and real discovery. A long candidate list is not the target. What matters is a report whose confidence level matches the evidence.
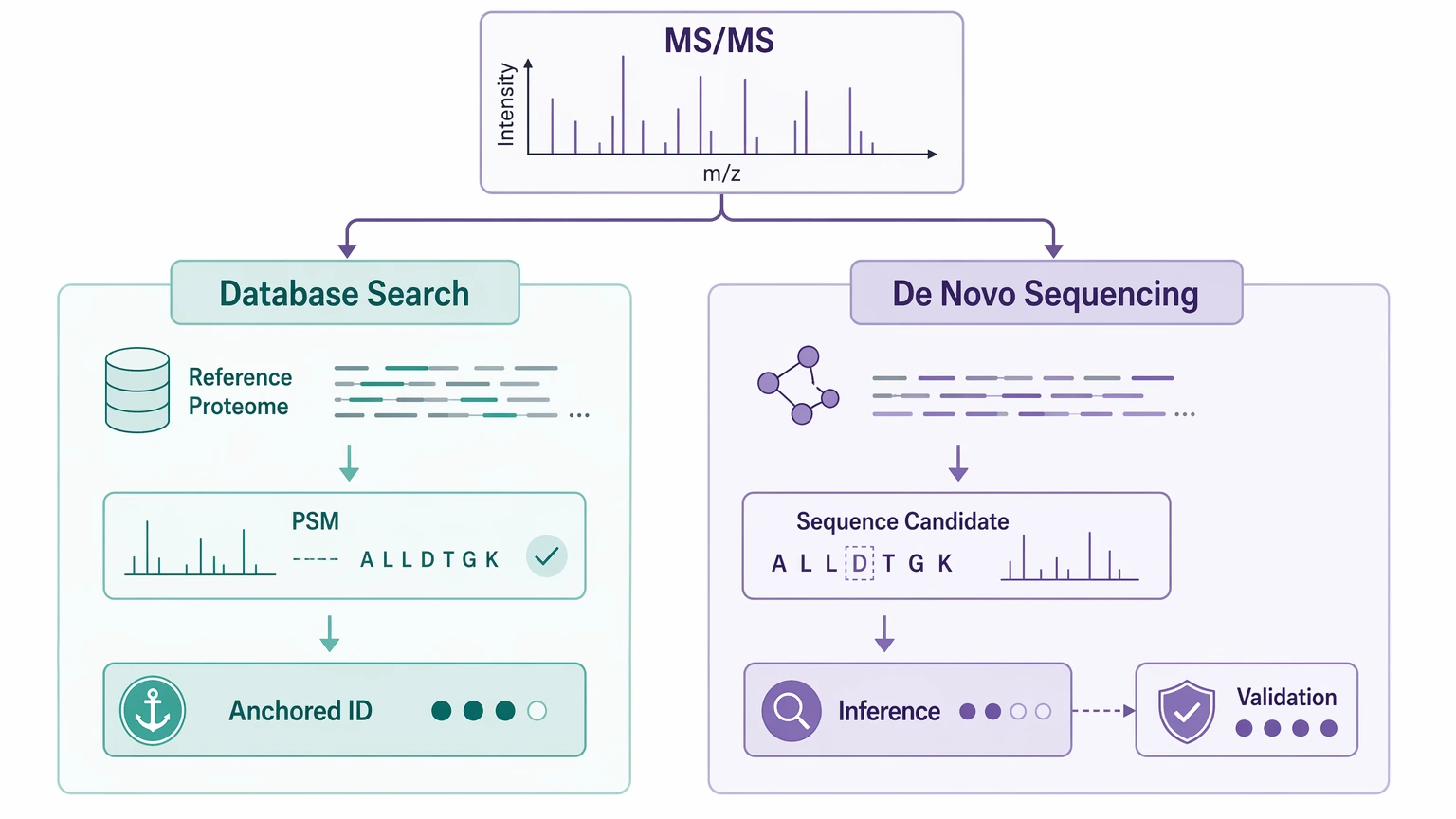
What the two workflows actually deliver
Database search compares experimental tandem mass spectrometry spectra with theoretical spectra generated from peptides already present in a searchable sequence resource. The output is usually a ranked set of PSMs, confidence scoring, and FDR-based filtering. It works well when the relevant peptide space is already represented in a high-quality reference proteome or custom database.
De novo sequencing infers peptide sequence candidates directly from fragment ions in the MS/MS spectrum. In bottom-up workflows, that usually means interpreting b ions / y ions or, depending on fragmentation mode, c ions / z ions. This route is most useful when the sequence is novel, the reference is incomplete, or the sample contains unexpected processing products.
The two outputs are not interchangeable. A strong PSM is a reference-anchored identification. A high-ranking de novo candidate is still an inference, and it often needs more support before it should be treated as established sequence evidence.
The four decision dimensions that matter most
Reference dependence versus sequence novelty
This is the main dividing line. If the biologically relevant peptide is absent, misannotated, or too divergent from the search database, database search may return no convincing hit or may promote a near match that looks computationally reasonable but does not fit the sample context. That risk gets higher in a low-reference organism, engineered constructs, microbiome-derived samples, venom peptides, and unusual cleavage products.
De novo sequencing stays useful when reference coverage breaks down because it does not require the correct sequence to be listed in advance. Even so, it still relies on interpretable spectra. It is not a rescue option for every unexplained scan.
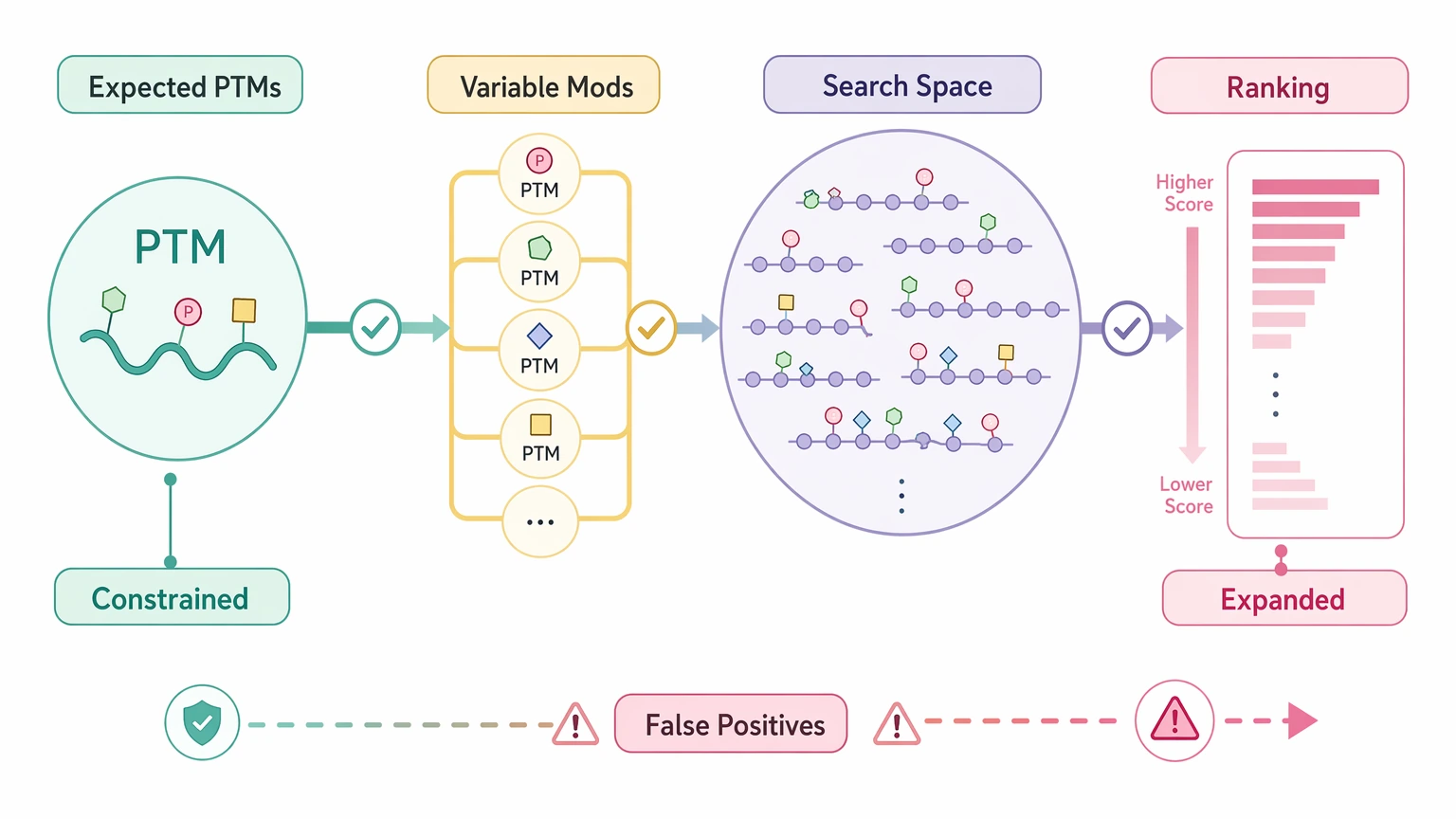
PTM burden and search-space inflation
A post-translational modification (PTM) can challenge both workflows, but not in the same way. Database search handles expected PTMs well when the modification list is biologically constrained. Once too many variable modification options are opened, the search space grows fast, ranking becomes less distinct, and false positives are harder to separate from real findings.
De novo sequencing can reveal unexplained mass shifts and local residue patterns around a modified region. That makes it useful for hypothesis generation, but it does not automatically resolve PTM localization. If site-defining fragment ions are sparse, the modified residue assignment may still remain uncertain.
Spectral quality and sequence coverage
De novo sequencing is far more sensitive to spectral quality than many routine search workflows. Better results usually require continuous ion evidence, strong precursor mass accuracy, strong fragment mass accuracy, and enough sequence coverage across the peptide to support residue-by-residue inference.
Database search is more forgiving when the correct sequence is already in the database. Partial ion series may still support a good PSM if the candidate space is narrow and the match is distinctive.
Reporting confidence and interpretation burden
Database search and de novo sequencing rest on different confidence logic. PSM-centric FDR frameworks fit database search well, but they do not transfer neatly to every form of de novo candidate ranking. That becomes important when teams want to claim sequence novelty from LC-MS/MS evidence alone.
One limitation should be stated clearly: standard bottom-up MS/MS often cannot fully resolve leucine/isoleucine ambiguity, and de novo sequence confidence may remain incomplete when fragmentation gaps, unexpected PTMs, or database-search limits leave more than one plausible interpretation.
When database search is the better first choice
Database search is usually the strongest starting point when three conditions are true:
In that setting, database search gives a clean first-pass view of what the data actually support. It also fits standard reporting practice for PSM review, confidence scoring, and replicate comparison.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
Still, database-only interpretation becomes weaker in three common situations:
When de novo sequencing becomes justified
De novo sequencing is justified when novelty is central to the project, not just a fallback after a disappointing search. Common examples include low-reference species, custom constructs, processed peptide mixtures, sequence-divergent samples, and projects where unexpected peptide content is biologically meaningful.
Its practical value has two sides. First, it can generate candidate sequences or partial sequence tags from high-information spectra. Second, those outputs can improve the downstream workflow, such as building a custom database, constraining re-search parameters, or linking peptide evidence to proteogenomics resources.
What de novo sequencing should not do is encourage over-interpretation. If the ion ladder is broken, residue evidence is weak, or multiple candidates fit the same spectrum, the right output may be a partial sequence tag rather than a final peptide claim.
Why hybrid workflows are often the safest choice
For many discovery projects, the safest route is neither database search alone nor de novo sequencing alone. A hybrid workflow often looks like this:
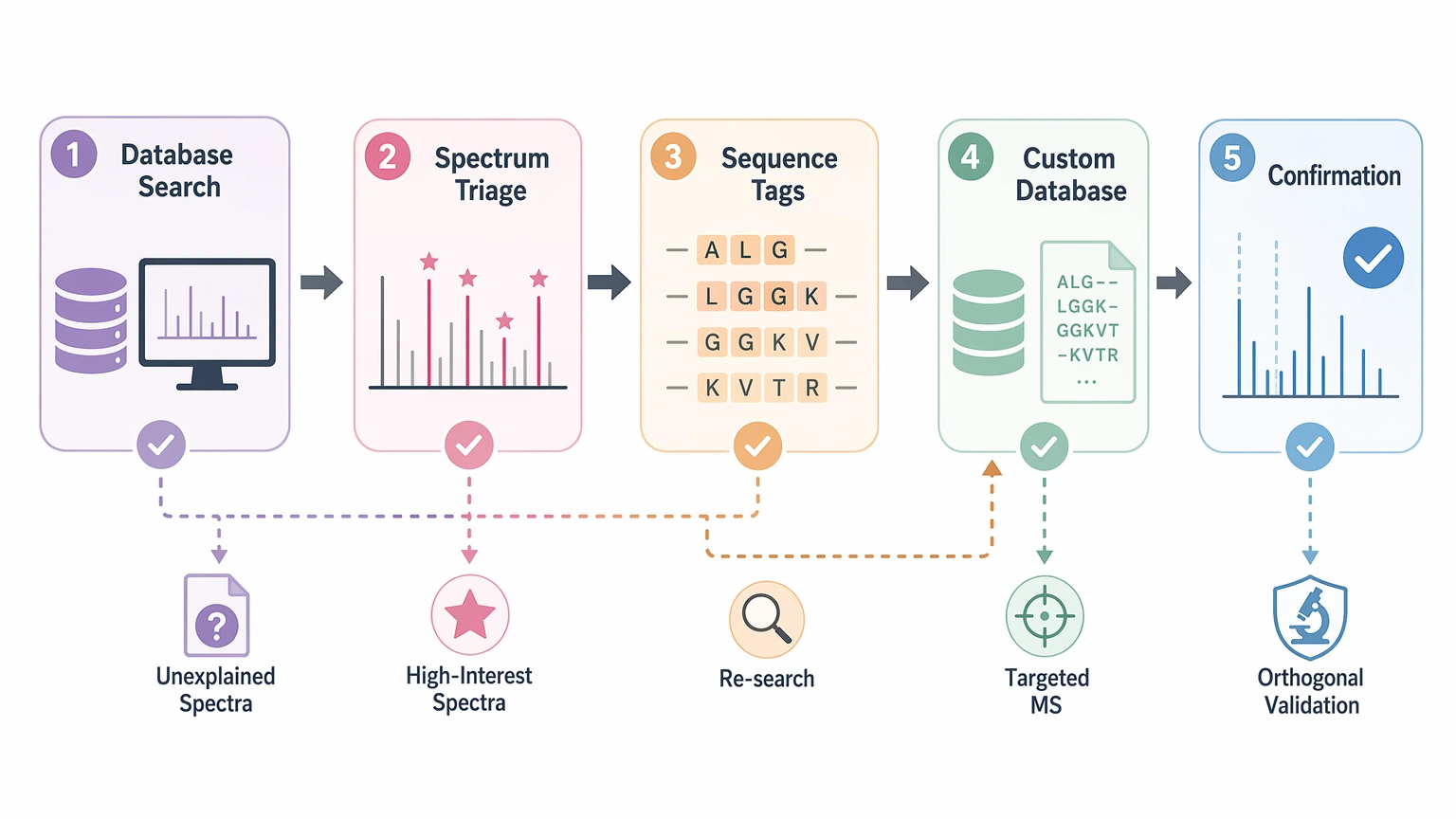
That structure helps separate three problems that often get mixed together:
If your team is at that point, submit your requirements or evaluate your project with MtoZ Biolabs around the LC-MS/MS data type, expected novelty, database limitations, and the level of confirmation needed before final reporting.
Expected results and validation methods
The right workflow should also be chosen with the final deliverable in mind.
| Evidence type | Immediate deliverable | What still needs follow-up confirmation |
|---|---|---|
| Database search result | Filtered PSM list with confidence scoring and FDR context | Manual review, replicate consistency, or targeted confirmation for critical claims |
| De novo sequence tag | Local residue pattern or partial candidate direction | Re-search against a custom database or confirm by targeted MS |
| Full de novo candidate sequence | Strong hypothesis for a novel peptide | Synthetic peptide confirmation, targeted LC-MS/MS, or proteogenomics support |
| PTM-associated mass shift | Candidate modified peptide region | Site-specific confirmation for PTM localization |
| Hybrid discovery result | Ranked candidates with reference and inference support | Orthogonal validation before strong novelty claims |
The distinction between immediate and follow-up outputs matters. A deliverable can still be analytically useful even when it is not yet biologically final.
Key cautions and practical limits
Before choosing a workflow, review the constraints that most often change interpretation quality:
A practical selection framework for live projects
Start with the claim your study needs to make. If your report only requires confident identification of expected peptides, database search is usually the right anchor. If the core question is whether the sample contains previously unlisted peptide content, de novo sequencing becomes more relevant. If the most important spectra look novel but still fall short of being definitive, a hybrid workflow is often safer than forcing certainty too early.
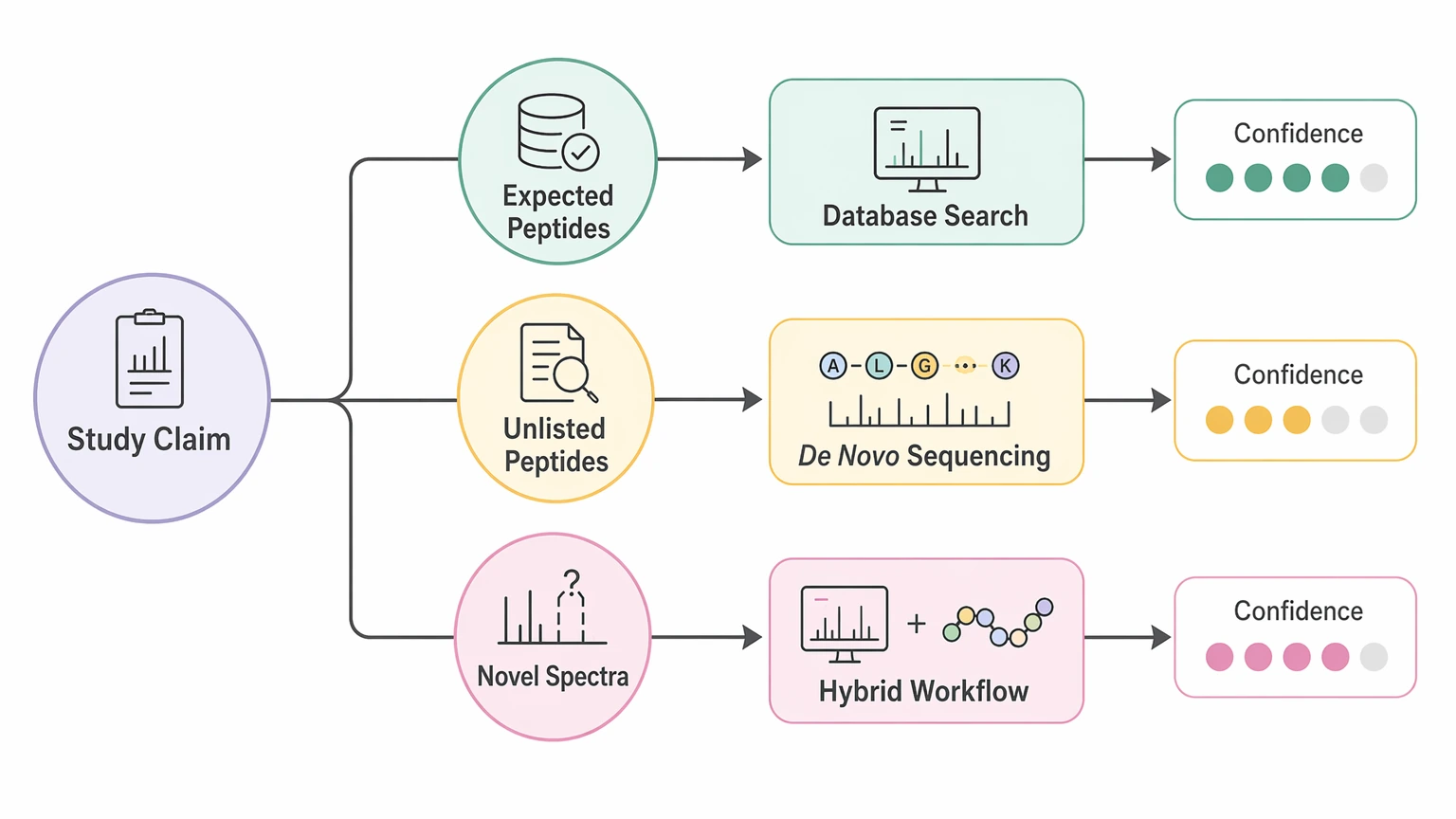
For studies with sparse reference coverage, unexpected processing products, engineered sequences, or PTM-rich discovery datasets, the next useful step is to define the sample source, expected peptide length range, fragmentation mode, database limitations, and validation endpoint before expanding the analysis. If you need to compare de novo sequencing, database search, or a staged hybrid route for an active dataset, contact MtoZ Biolabs to discuss the workflow, evaluate your project, and align the report format with the level of sequence confidence your team needs.
FAQ
Can a custom database remove the need for de novo sequencing?
Sometimes, but only if you can populate that custom database with biologically plausible candidates. If the unknown sequence space is still broad, de novo-derived sequence tags may be what makes the custom database useful in the first place.
Which fragmentation modes make de novo sequencing more informative?
Modes that produce richer and more continuous fragment-ion series usually help. In practice, the best choice depends on peptide class, charge state, and whether PTM retention during fragmentation is a major concern.
Should every unexplained spectrum be sent into de novo analysis?
No. Start with spectra that are high quality, biologically relevant, and not already explained by reasonable database-search hypotheses. Poor spectra can generate misleading de novo candidates.
How much novelty evidence is enough for publication-ready reporting?
That threshold depends on the claim. A candidate sequence may be suitable for internal prioritization, while external reporting of a novel peptide often needs synthetic peptide confirmation, targeted follow-up, or supporting genomic or transcriptomic evidence.
Does FDR work the same way for de novo sequencing?
Not exactly. Database-search FDR is centered on PSM competition in a defined search space. De novo confidence scoring reflects inference quality, and its relationship to final sequence correctness needs to be explained more carefully.
What should be prepared before outsourcing workflow selection?
Prepare the organism or construct context, raw LC-MS/MS files, current search results, expected peptide length range, suspected PTMs, and the level of downstream confirmation your project can support.
How to order?

