





De Novo Sequencing of Monoclonal Antibodies: Planning a Sequence Confirmation Strategy for Therapeutic mAbs
- Use database-based peptide mapping alone when the reference sequence is trusted and the goal is routine identity support.
- Add de novo sequencing of monoclonal antibodies when you need to test undocumented substitutions, incomplete records, or weak CDR assignments.
- Add orthogonal confirmation when a disputed site could change a comparator conclusion, sequence verification package, or CMC handoff decision.
- Treat any apparent difference as provisional until overlapping peptide evidence, PTM-aware review, and chain-level context support the same call.
- what decision will the final report support
- which chain, region, or residue carries the highest project risk
- what level of unresolved ambiguity is still acceptable
- whether the sample is purified mAb, formulated product, reduced chains, or subunits
- approximate amount available for repeat digestion or targeted reanalysis
- whether visible degradation, carryover risk, or mixed material is already suspected
- whether the main concern sits in the variable region, framework region, termini, hinge, or glycosylated region
- separate review of heavy chain and light chain coverage maps
- multi-enzyme digestion to produce overlapping sequence tags
- LC-MS/MS settings suitable for challenging peptide classes
- region-specific review of CDRs, termini, hinge peptides, and disulfide-constrained peptides
- PTM-aware interpretation so modified peptides are not misread as substitutions
- each CDR, especially CDR3
- N-termini and C-termini
- hinge and hinge-adjacent peptides
- glycosylation-bearing segments
- disulfide-rich regions that may need disulfide mapping
- any site with a suspected amino acid substitution
- positions affected by leucine/isoleucine ambiguity
- a heavy chain and light chain coverage map
- confidence-ranked de novo peptide assignments
- a list of unresolved residues or low-confidence positions
- a summary of PTM-related interpretation conflicts
- chain- and region-level comments on variable region and framework region support
A credible sequence confirmation strategy for a therapeutic monoclonal antibody starts with a focused question: what evidence is strong enough to confirm, challenge, or leave unresolved the expected heavy chain and light chain sequence? In many therapeutic mAb programs, routine peptide identification alone does not settle that question. The more practical standard is residue-level confidence in the regions that affect development decisions, especially each complementarity-determining region (CDR), termini, hinge-adjacent peptides, and peptides complicated by post-translational modification (PTM).
Quick decision guide
If an expected sequence already exists, the real task is to separate confirmed positions from uncertain ones. The same logic applies when reviewing originator, biosimilar, or legacy material: an observed difference is not yet a sequence variant unless the fragmentation evidence is stronger than the reasonable analytical alternatives.
Define the sequence confirmation problem before choosing the workflow
Teams often say they need to “confirm the antibody sequence,” but that phrase can point to very different decisions. One program needs independent sequence verification before a milestone. Another needs to resolve a suspected amino acid substitution in a comparator study. A third needs stronger confidence in variable region assignments before tech transfer or documentation.
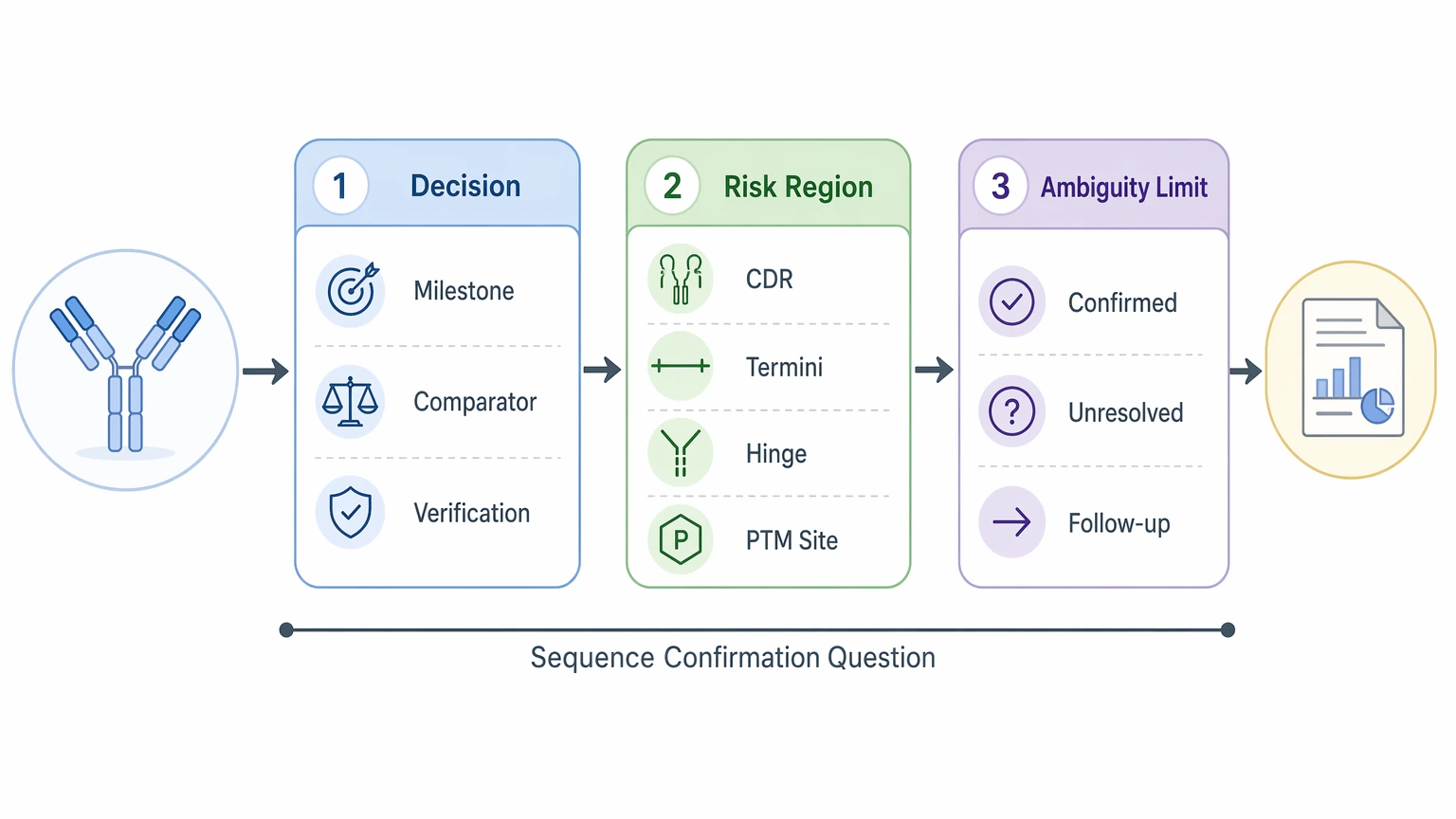
That distinction matters because the evidence bar changes with the decision. If the concern is one CDR3 position, broad overall coverage helps, but targeted support for that local region matters more. If the question is whether two materials are sequence-matched at the protein level, heavy chain and light chain assignments, overlapping peptides, and PTM-aware review matter more than a single high-level coverage metric.
A planning discussion should therefore answer three questions up front:
Why standard workflows leave uncertainty in therapeutic mAbs
For this topic, four root causes usually explain why sequence confidence stops short.
1. The project goal is broader than the data package
A generic “sequence confirmation” request often leads to a generic dataset. That rarely works well when the actual decision turns on one variable region, a disputed terminus, or a suspected substitution. Therapeutic mAbs need a workflow aligned to the highest-risk region, not just whole-molecule coverage.
2. One digestion route rarely covers all regions well
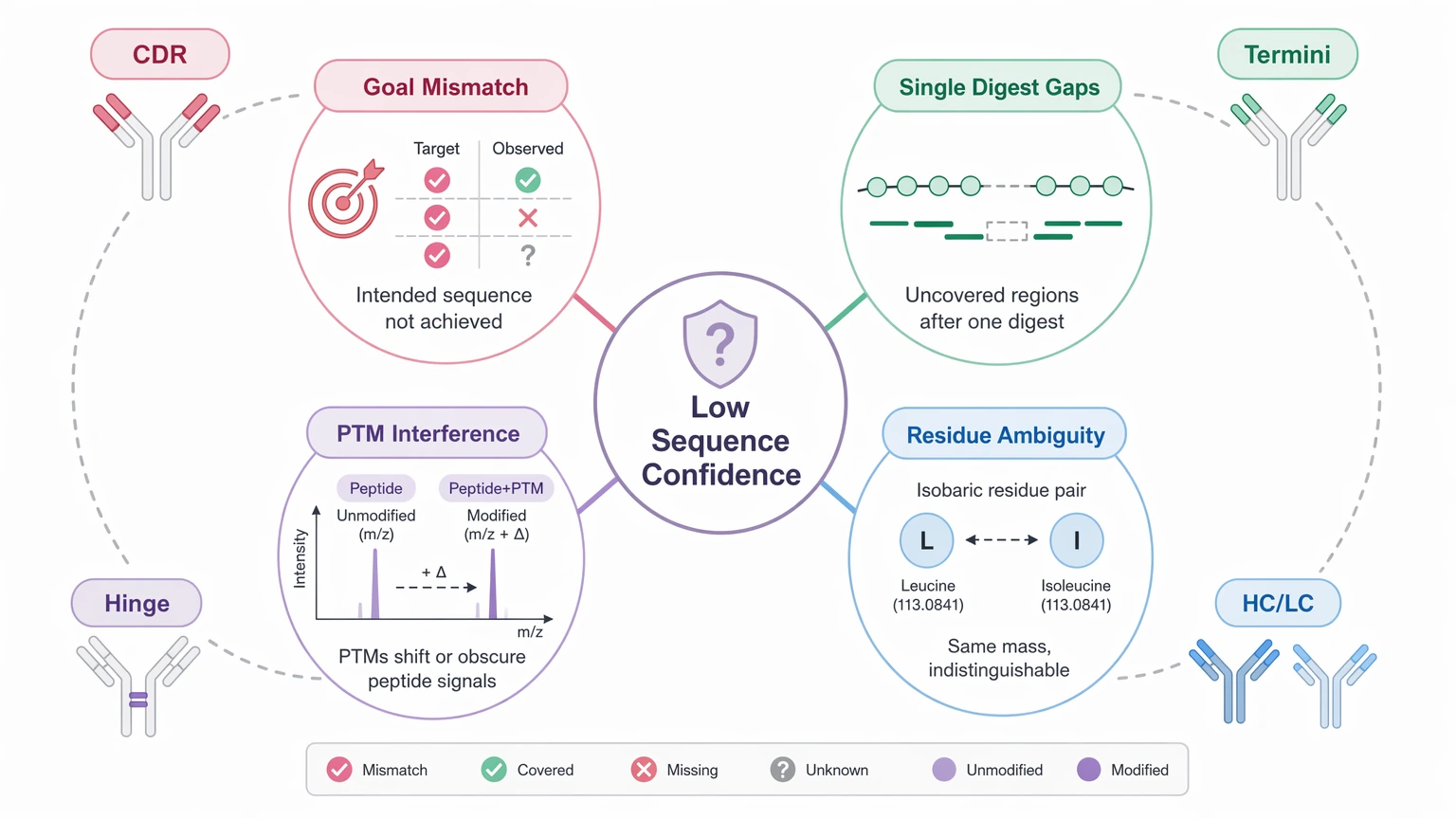
A single protease can leave long peptides, short peptides, missed cleavages, or poorly fragmented segments. Disulfide-constrained and hinge-adjacent peptides are common weak points. So a coverage map may look acceptable while the most important local assignments are still thin.
3. PTMs can obscure sequence interpretation
Glycosylation, oxidation, deamidation, pyroglutamate formation, and glycation can shift mass or change fragmentation behavior. In difficult peptides, a PTM can look like an amino acid substitution or lower confidence in a sequence tag. That is why PTM-aware interpretation belongs inside the sequencing workflow rather than as an afterthought.
4. Residue-level ambiguity is sometimes overcalled
Leucine/isoleucine ambiguity is the most familiar example because those isobaric residues cannot always be separated by standard LC-MS/MS evidence alone. Low-intensity spectra, uneven overlap across digests, and uncertain heavy chain versus light chain assignment can create the same problem. A credible report should label those positions clearly instead of forcing a full call. In de novo peptide interpretation, MS/MS-based sequence confidence can remain limited at specific residues even when the surrounding peptide assignments are strong.
Project-planning workflow for de novo sequencing of monoclonal antibodies
Step 1: Tie the workflow to the decision question
Write the project aim as a testable statement. Examples include confirming the expected heavy chain and light chain sequence, checking whether a suspected substitution is real, or improving confidence in complementarity-determining region assignments before a handoff package. This first step sets the boundary for what follow-up work is worth doing.
A quick planning table can help match the workflow to the decision.
| Scenario | Recommended workflow | Main limitation | Preferred follow-up |
|---|---|---|---|
| Known reference, routine confirmation | Multi-enzyme digestion plus targeted de novo review of weak peptides | Isolated low-confidence residues may remain | Recheck disputed sites only |
| Suspected sequence variant | Overlapping de novo peptide interpretation with PTM-aware review | Single-peptide differences are not strong evidence | Targeted site confirmation |
| Comparator study | Intact mass analysis, subunit analysis, and peptide-level sequence verification | PTMs can mimic sequence differences | Alternate digest or targeted LC-MS/MS |
| Incomplete legacy record | Broad heavy chain and light chain de novo sequencing | Chain assignment may remain uneven in difficult regions | Terminal or transcript cross-check |
Use the table to scope the decision, not just the instrument time. The right package is the one that answers the specific risk question with the fewest unsupported assumptions. If your team needs help matching sample format, risk regions, and deliverables before submission, you can submit your requirements to MtoZ Biolabs to evaluate the project scope around antibody sequencing and targeted confirmation.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
Step 2: Screen sample suitability early
Sample form shapes the kind of evidence you can realistically expect. Purified monoclonal antibody material is usually the clearest starting point. Formulated drug product may still work, but excipients can complicate digestion or reduce spectral quality. Reduced chains or Fab/Fc subunits can improve local interpretation, although they also remove some intact context.
Check these points before locking the workflow:
This step keeps the plan realistic. A stressed or matrix-heavy sample can still be useful, but the final claims may need tighter limits.
Step 3: Design for overlapping evidence, not just broad coverage
In therapeutic mAb sequence verification, confidence improves when multiple peptides support the same local assignment. That usually means multi-enzyme digestion plus complementary tandem mass spectrometry behavior across difficult peptides. Framework region peptides often confirm readily, but variable region peptides, termini, and modified segments need more deliberate redundancy.
A stronger design typically includes:
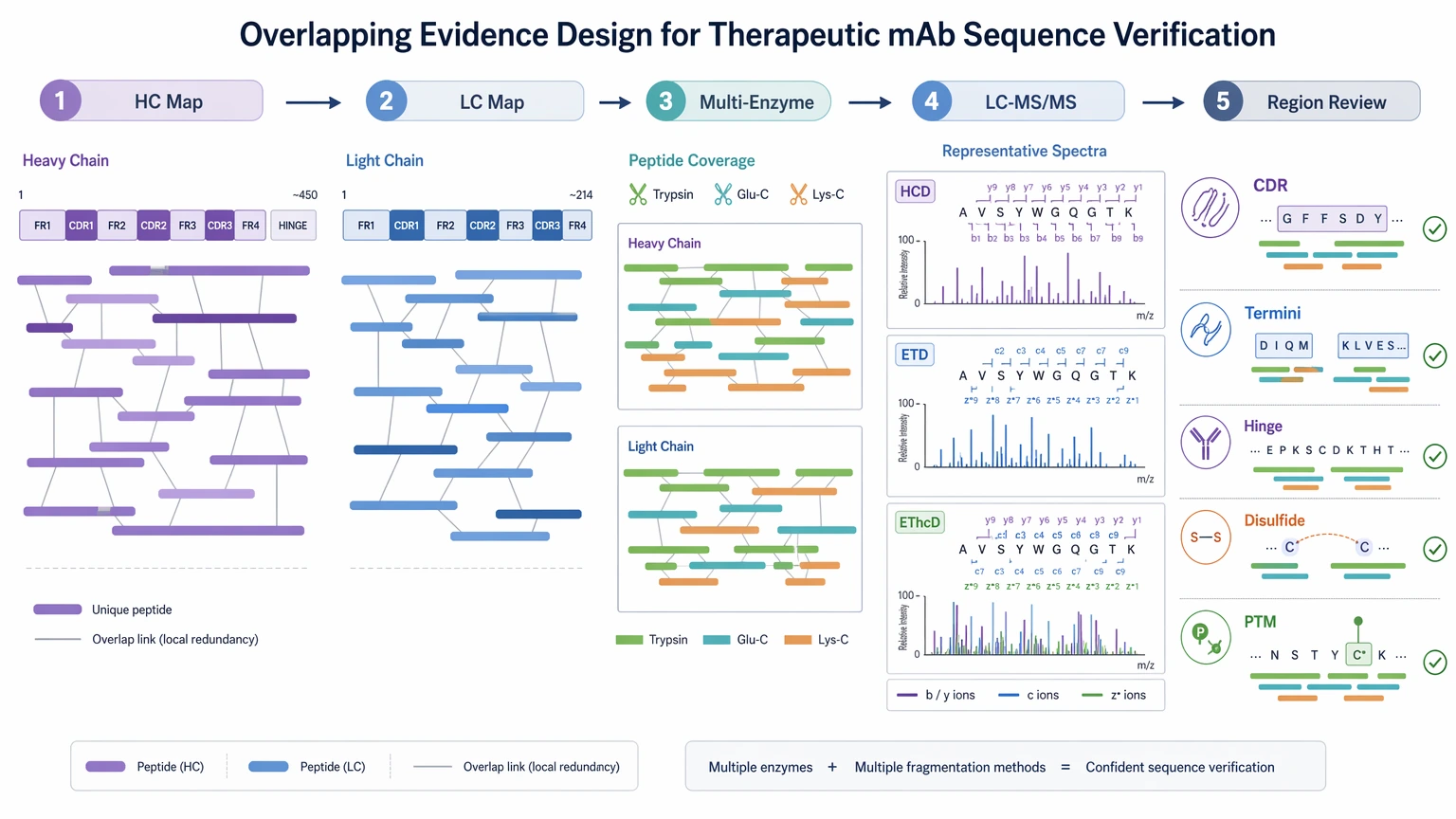
For difficult sites, complementary fragmentation such as HCD together with ETD or EThcD can improve local interpretation. Even then, no single fragmentation mode resolves every peptide equally well, and some assignments still need follow-up confirmation.
Step 4: Predefine ambiguity-prone regions
Do not wait until final reporting to decide which residues matter most. Flag high-risk regions before analysis so the team knows where extra review is likely.
Priority regions usually include:
For each of those regions, define what will count as acceptable support. That might mean overlapping peptides from more than one digest, consistent chain assignment, or agreement between peptide mapping and de novo sequencing.
Expected results and validation methods
A well-planned workflow does not produce certainty at every residue. The immediate deliverables are usually:
Those outputs form the working evidence package. Follow-up confirmation is a separate step. It may include targeted LC-MS/MS reanalysis, alternate protease digestion, subunit analysis, intact mass analysis, disulfide mapping, terminal sequencing, or transcript-derived sequence cross-checking.
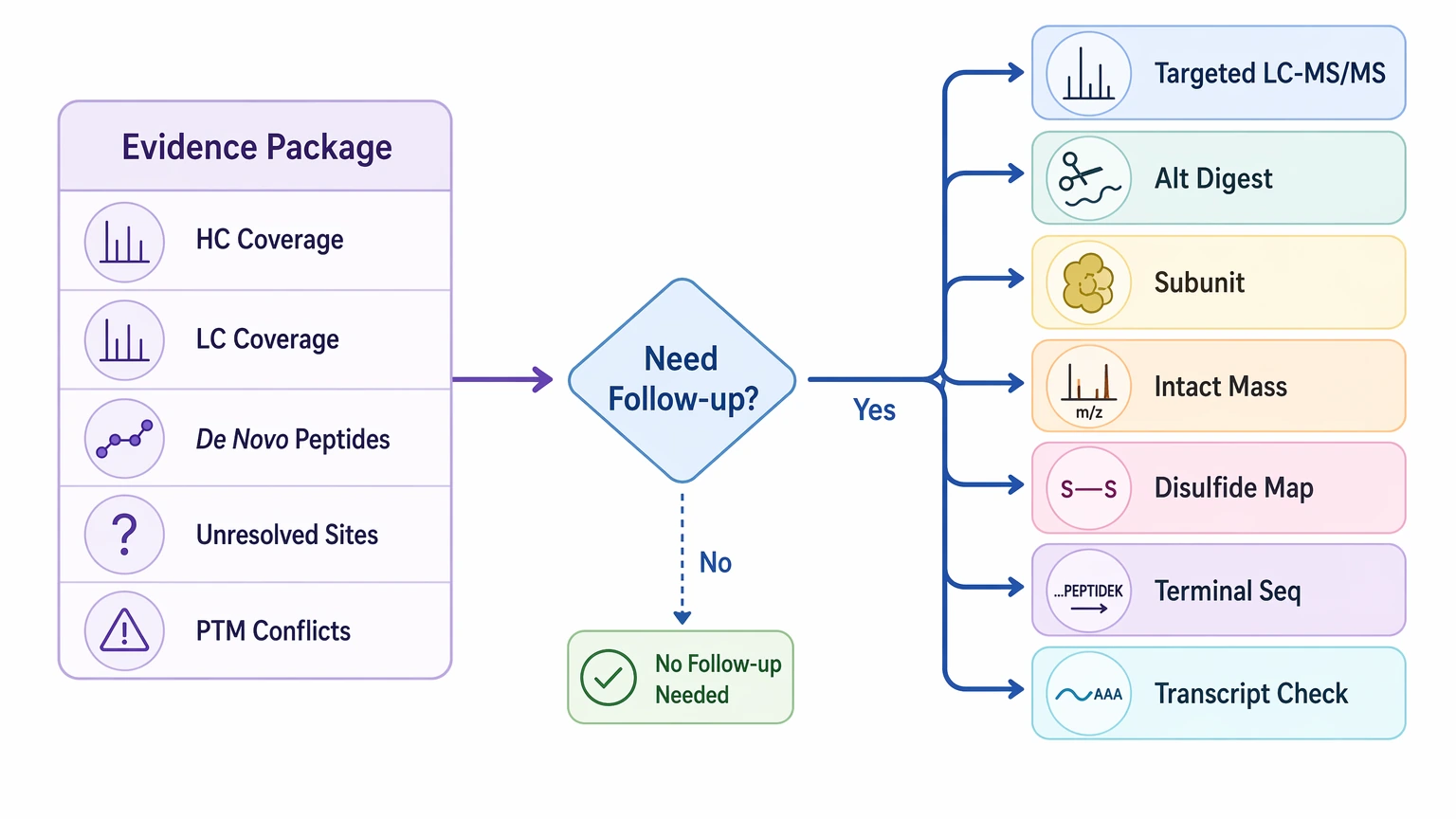
That distinction matters. Immediate deliverables show where the data are strong, weak, or conflicting. Follow-up confirmation tests whether the unresolved positions matter enough to change the project decision. In comparator work, that is often the point where a provisional difference becomes either a supported sequence variant or a likely analytical artifact.
Key cautions and practical limits
Several limits should stay explicit in the report and in internal discussions.
First, sample quality and amount place real boundaries on interpretation. Degraded, low-purity, or matrix-heavy samples narrow the useful evidence space and may reduce repeat options.
Second, controls and repeat logic should be planned before claims are made. If the decision depends on one substitution call, repeat evidence or orthogonal confirmation is usually more useful than broader but redundant coverage.
Third, batch effects, contamination, and mixed material can create false discrepancy signals. A comparator study is especially vulnerable if lot-specific modification differences are mistaken for sequence differences.
Fourth, interpretation has clear boundaries. A broad coverage map does not prove every residue, and a single peptide does not settle every disputed position. Leucine/isoleucine ambiguity may remain unresolved without outside evidence, and PTM-heavy peptides can still limit local sequence confidence.
Finally, another method may be the better next step when the core question is very narrow. If the project centers on one terminus, one unresolved isobaric residue, or one highly modified peptide cluster, targeted validation or transcript-based cross-checking may be more efficient than expanding a full de novo workflow. If that decision point is close, contact MtoZ Biolabs to discuss the sample scenario, review likely blind spots, and evaluate your project before committing additional material.
Conclusion
For a therapeutic monoclonal antibody, a credible sequence confirmation strategy usually combines de novo sequencing, peptide mapping, chain-aware interpretation, and selective orthogonal confirmation rather than leaning on one readout alone. That approach fits therapeutic mAb sequence verification, comparator review, and milestone-stage record checks when the key question is whether protein-level evidence supports the expected heavy chain and light chain sequence with clear confidence boundaries. If your project involves uncertain CDR assignments, a suspected sequence variant, or limited sample that needs careful prioritization, prepare the sequence record, sample format, known PTM concerns, and the exact decision the data must support before requesting a feasibility review.
FAQ
When does de novo sequencing add more value than routine peptide mapping?
It adds value when the expected sequence is incomplete, a comparator result shows a mismatch that database search does not explain, or the decision depends on direct peptide-level evidence in a variable region.
How should heavy chain and light chain evidence be reported?
They should be reported separately first, then integrated. Separate reporting shows whether one chain has weaker coverage, lower sequence confidence, or more unresolved residues than the other.
What is enough evidence to call a real sequence variant?
A real call usually needs overlapping peptide support, consistent chain assignment, review against plausible PTMs, and confirmation that the difference is reproducible across a second analytical angle.
Are CDRs always the hardest regions to confirm?
Not always. CDRs are high priority because of biological relevance, but termini, hinge-adjacent peptides, glycosylated regions, and disulfide-constrained peptides can be just as difficult analytically.
Should transcript-derived sequence data replace protein-level confirmation?
No. Transcript data can help resolve disputed positions, but it does not replace protein-level evidence for processing, PTMs, chain assignment, or product-form differences.
What should be included in a sequencing feasibility request?
Include the expected sequence if available, sample type, approximate amount, formulation or purity concerns, suspected discrepancy sites, comparator context, and the internal decision that the evidence package must support.
How to order?

