





De Novo Sequencing Methods in Proteomics: Where MS-Based Approaches Add the Most Value
- De novo sequencing infers amino acid order directly from a tandem mass spectrometry dataset rather than relying only on a database search or spectral library search.
- It adds the most value when the expected sequence is missing, incomplete, highly variable, or biologically processed in an unexpected way.
- Strong peptide de novo sequencing depends on interpretable b ions, y ions, precursor isolation quality, fragment mass accuracy, and manageable spectral complexity.
- Protein de novo sequencing is usually harder than peptide-level reconstruction because it may require multiple peptides, terminal information, or support from top-down proteomics.
- Many useful outputs are partial rather than absolute: a sequence tag, a ranked candidate list, PTM-aware hypotheses, or a clear explanation of why current data do not support higher sequence confidence.
- a full peptide sequence for the best-supported analyte
- one or more partial sequence tags
- ranked candidate sequences with ambiguity notes
- PTM annotations and provisional PTM localization
- spectrum-level evidence summaries for manual spectrum interpretation
MS-based de novo sequencing is most useful when the main obstacle is sequence novelty, not routine identification sensitivity. In practical terms, the target peptide or protein may be missing from available references, poorly annotated, unexpectedly processed, or modified in a way that weakens a standard database search. If the LC-MS/MS data also contain clear fragment-ion evidence, de novo interpretation can yield full peptide sequences, informative sequence tags, or ranked candidate sequences that help move the study forward.
In most projects, the real choice is not “database search only” versus “pure de novo” as two fixed camps. Proteomics teams usually get better answers by matching the workflow to the source of uncertainty: known target space, partly known target space, or genuinely unknown sequence space. Sample purity, precursor ion isolation quality, peptide length, fragmentation method, and the feasibility of orthogonal validation usually matter more than the software label.
Quick Decision Guide
Use database search first when the organism, construct, or protein family is already well represented in reference databases.
Use a hybrid workflow when reference coverage is incomplete, but partial sequence context still exists.
Move to de novo sequencing when unknown peptide identification or novel sequence identification is the main goal and the MS/MS spectrum quality is strong enough to support direct residue inference.
Do not expect every residue to be final from MS/MS alone: Leucine/Isoleucine ambiguity, post-translational modification (PTM) complexity, and incomplete fragment ladders can leave more than one plausible sequence interpretation.
Key Takeaways
What De Novo Sequencing Means in Proteomics
In proteomics, de novo sequencing means deriving sequence information directly from an MS/MS spectrum generated in an LC-MS/MS experiment. Instead of asking whether an observed spectrum matches a known database entry, the workflow reads mass differences between fragment ions to infer residue order.
For peptide de novo sequencing, the most familiar evidence comes from complementary b ions and y ions produced under CID or HCD, with ETD often added when charge state or PTM behavior makes collision-based fragmentation less informative. A clean, continuous fragment series can support direct peptide reconstruction. A broken or sparse ladder may still support a shorter sequence tag that helps with downstream searching or validation.
For protein de novo sequencing, the scope is broader. Bottom-up peptide evidence may need to be combined with terminal information, intact-mass context, or proteoform-level evidence from top-down proteomics. That is why protein-level sequence claims usually require more integration and more cautious interpretation than a strong single-peptide result.
Why Database-Dependent Workflows Sometimes Stop Short
Reference-driven methods remain the default for good reason. A database search is efficient when the correct sequence is already represented, and a spectral library search can be even stronger when relevant reference spectra are available. Problems start when those assumptions no longer hold.
The clearest failure mode is missing reference content. Non-model organisms, poorly annotated strains, venom peptides, natural products, engineered constructs, and proprietary biologics may simply lack the right entries in public databases. In that situation, a weak search result does not always mean the analyte is absent. Sometimes the correct sequence was never in the search space.
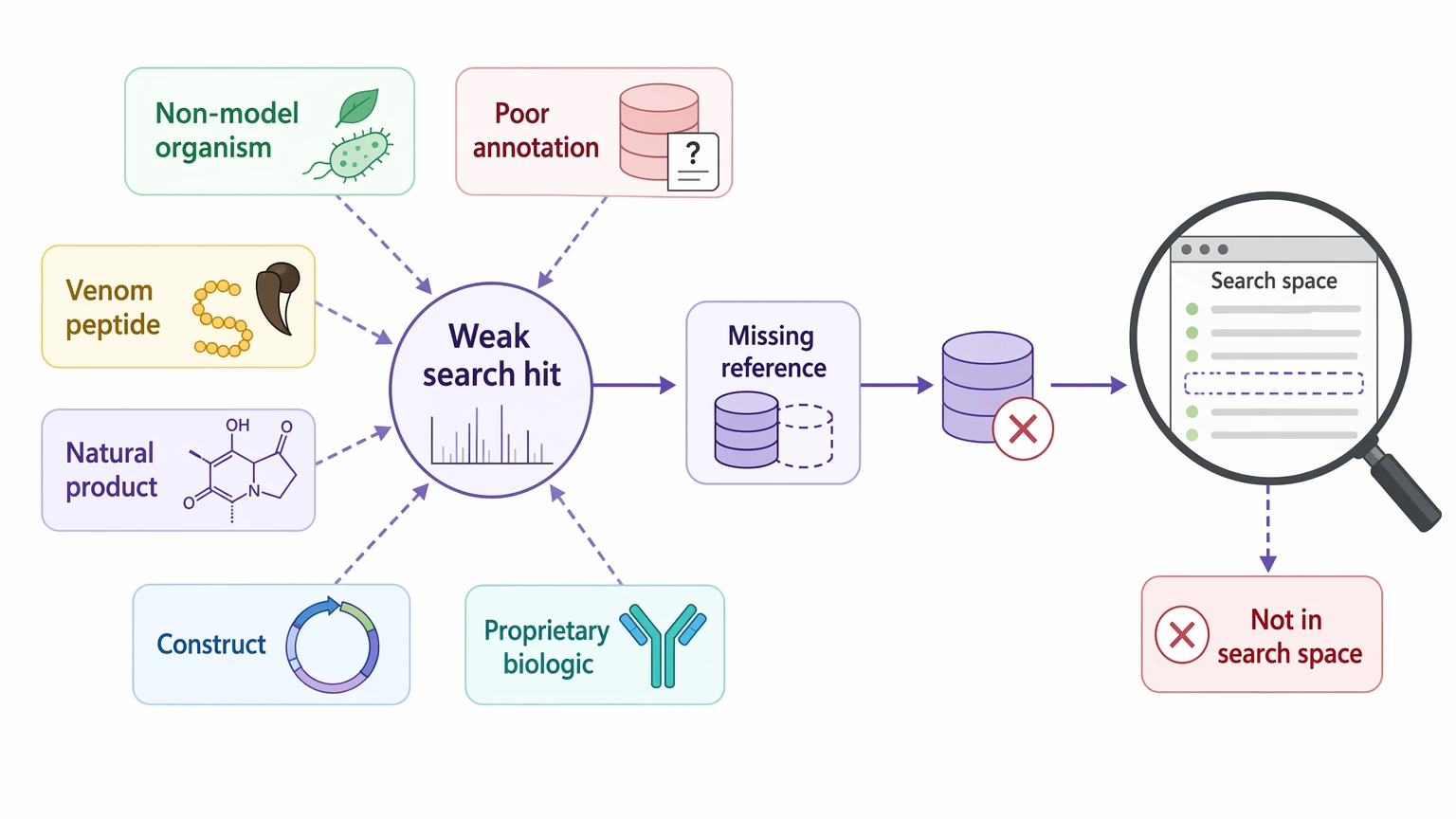
A second failure mode is unexpected biological processing. Cleavage products, terminal trimming, antibody-derived fragments, and noncanonical sequence variants may differ enough from the expected model to break routine matching. A third is modification burden. Phosphorylation, glycosylation, oxidation, and deamidation can shift precursor mass, change fragment-ion patterns, and complicate PTM localization. Under those conditions, reference-based scoring may favor incomplete or misleading explanations.
Where MS-Based De Novo Sequencing Adds the Most Value
The highest-value use cases are the ones where direct sequence inference answers a question that reference-driven methods cannot resolve cleanly.
First, de novo sequencing fits unknown peptide identification well. If a team is isolating an unannotated signaling peptide, a venom component, or a novel bioactive fragment, the main need is direct sequence evidence. Even a partial sequence tag can sharply narrow the next experiment.
Second, it helps with novel sequence identification in database-poor systems. Species or strain variants, engineered peptides, and proprietary constructs often sit in the middle ground between “known enough for routine search” and “fully unknown.” In these studies, a hybrid workflow is often the better choice: use database search for conserved regions, then use de novo sequencing to recover unsupported or divergent segments.
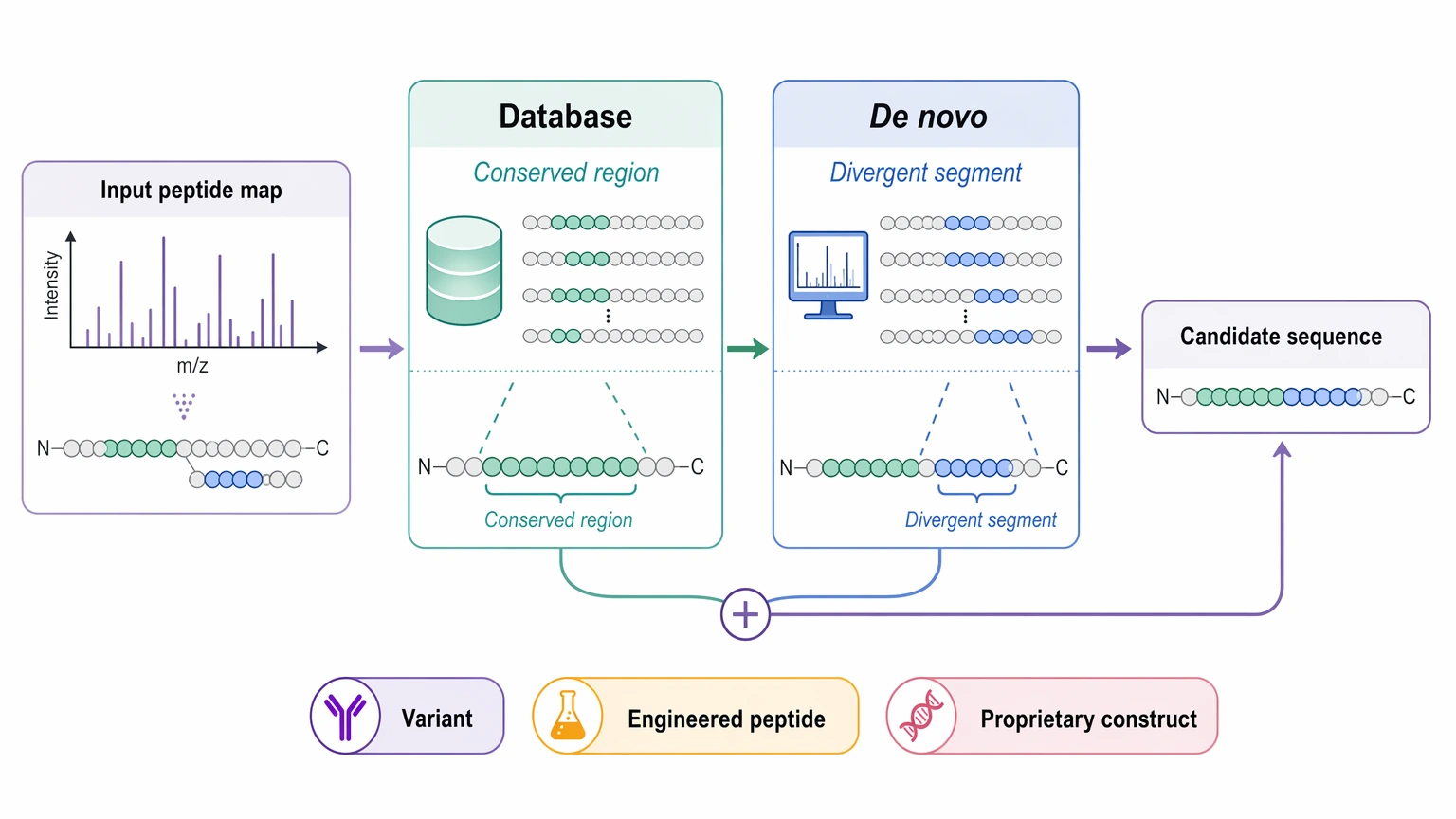
Third, de novo interpretation becomes especially useful when PTMs are central to the biology rather than a side detail. A modified peptide may still be identifiable by reference search, but if the study depends on site-specific reasoning or on separating competing modified sequence models, direct fragment interpretation becomes more informative. At the same time, PTM-rich analytes also raise uncertainty. A PTM can justify de novo analysis while also lowering sequence confidence by changing fragmentation behavior or leaving localization only partially ranked.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
A practical decision point often comes up here. When teams already have raw LC-MS/MS data from an unknown-sequence project and need to judge whether deeper de novo analysis is realistic, they can submit your requirements and evaluate your project with MtoZ Biolabs based on sample type, fragmentation quality, expected deliverables, and validation goals before committing more sample.
What Makes a De Novo Result Realistic
The strongest predictors are spectral interpretability and project context.
A clean precursor ion helps because the resulting MS/MS spectrum is more likely to represent one dominant analyte. Lower co-isolation burden means less mixed-fragment interference and a lower false assignment risk. Fragment ladders matter just as much. Longer runs of assigned b ions and y ions improve continuity and make candidate ranking easier to defend.
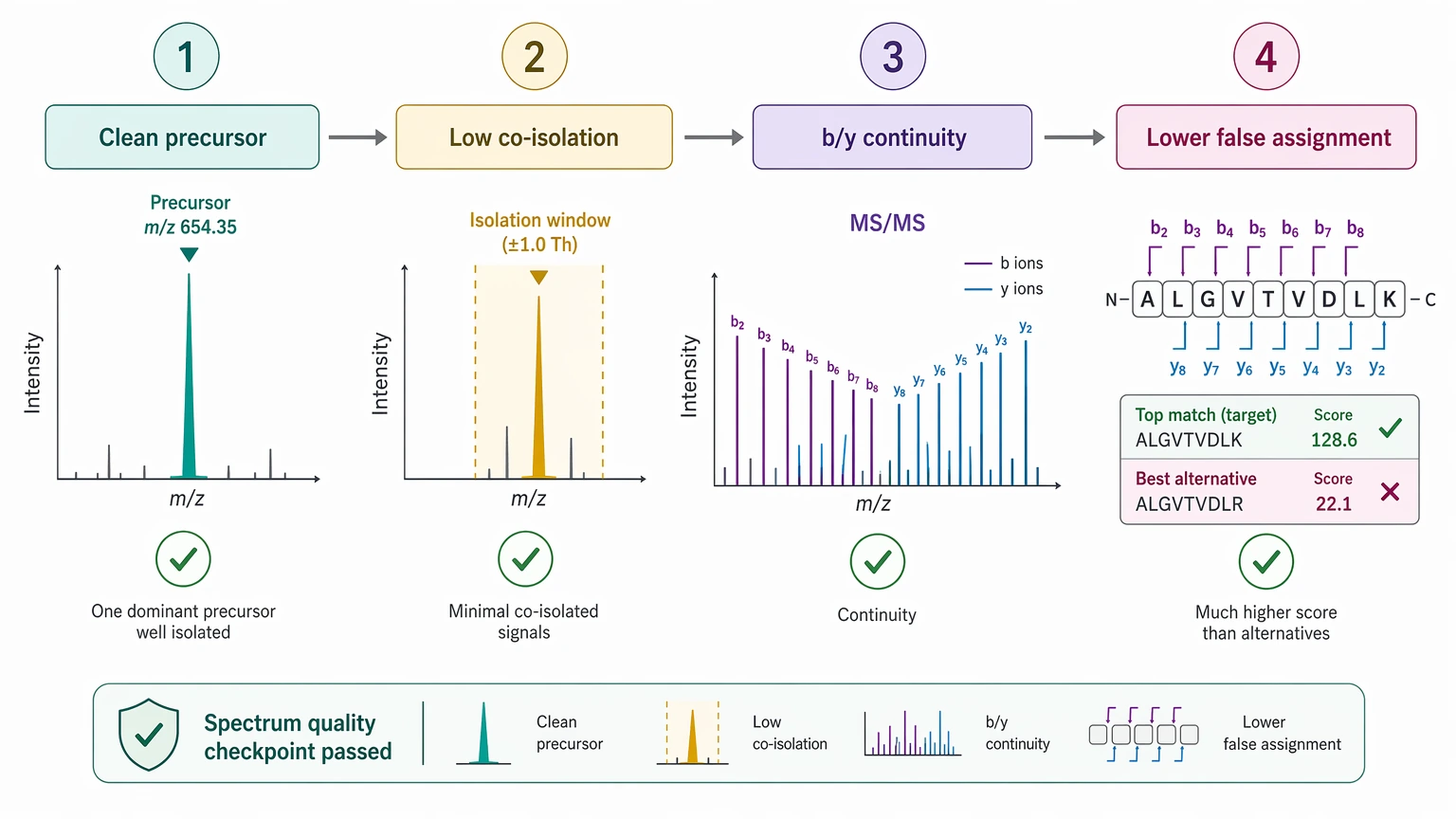
Fragmentation method choice also changes the outcome. HCD often gives useful backbone coverage for many peptides. CID still supports strong manual and algorithmic interpretation in established workflows. ETD becomes especially relevant when collision-based methods do not preserve enough useful backbone information, as in some highly charged or labile PTM-bearing peptides.
Mass accuracy improves discrimination, but it does not remove every ambiguity. Isobaric amino acids remain a basic limitation, and Leucine/Isoleucine ambiguity often cannot be resolved by standard MS/MS alone. Database-search limits can also remain after de novo reconstruction if the inferred sequence still has to be mapped back to a protein origin in an incomplete reference set.
Expected Results and Validation Methods
Before starting, teams should align on what the workflow is likely to return.
Immediate deliverables from de novo sequencing often include:
These outputs are analytically useful, but they are not always the final biological proof point.
Follow-up confirmation adds the next layer. Depending on the project, this can include targeted LC-MS/MS of the proposed sequence, synthetic peptide matching, terminal sequencing, transcriptomic or genomic cross-checking, or additional top-down support for a related proteoform question. If a decision depends on exact residue identity, exact PTM placement, or protein-level origin, orthogonal validation should be planned early rather than treated as an optional add-on.
Key Cautions and Practical Limits
Several constraints should shape project expectations.
Sample quality or amount limits: low-abundance targets, contaminated preparations, or limited material can restrict repeat acquisition and reduce fragment coverage.
Controls and repeat expectations: a single promising spectrum may be enough to form a hypothesis, but confirmation usually benefits from replicate acquisition, targeted follow-up, or an independent validation route.
Batch and contamination risk: mixed peptide populations, carryover, and precursor co-isolation can create composite spectra that look cleaner than they really are.
Interpretation boundaries: de novo outputs can support strong sequence hypotheses, but they do not automatically establish complete biological identity, exact protein origin, or universal PTM certainty. This matters most when the project involves sparse MS/MS evidence, multiple PTMs, or database-poor species.
When another method is the better next step: if the main question is routine protein identification in a well-annotated system, a database search remains the better first step. If terminal structure is the core unknown, terminal sequencing may be more direct. If intact proteoform context is central, a top-down strategy may answer the question more clearly than bottom-up de novo alone.
Choosing the Right Workflow for Project Planning
A useful planning question is simple: what exactly is blocking identification? If the main issue is missing reference content, de novo sequencing becomes more attractive. If the issue is only suboptimal search settings in a well-annotated system, stronger reference-driven analysis may solve it faster. If the answer falls somewhere in between, a hybrid workflow is often the most efficient route.
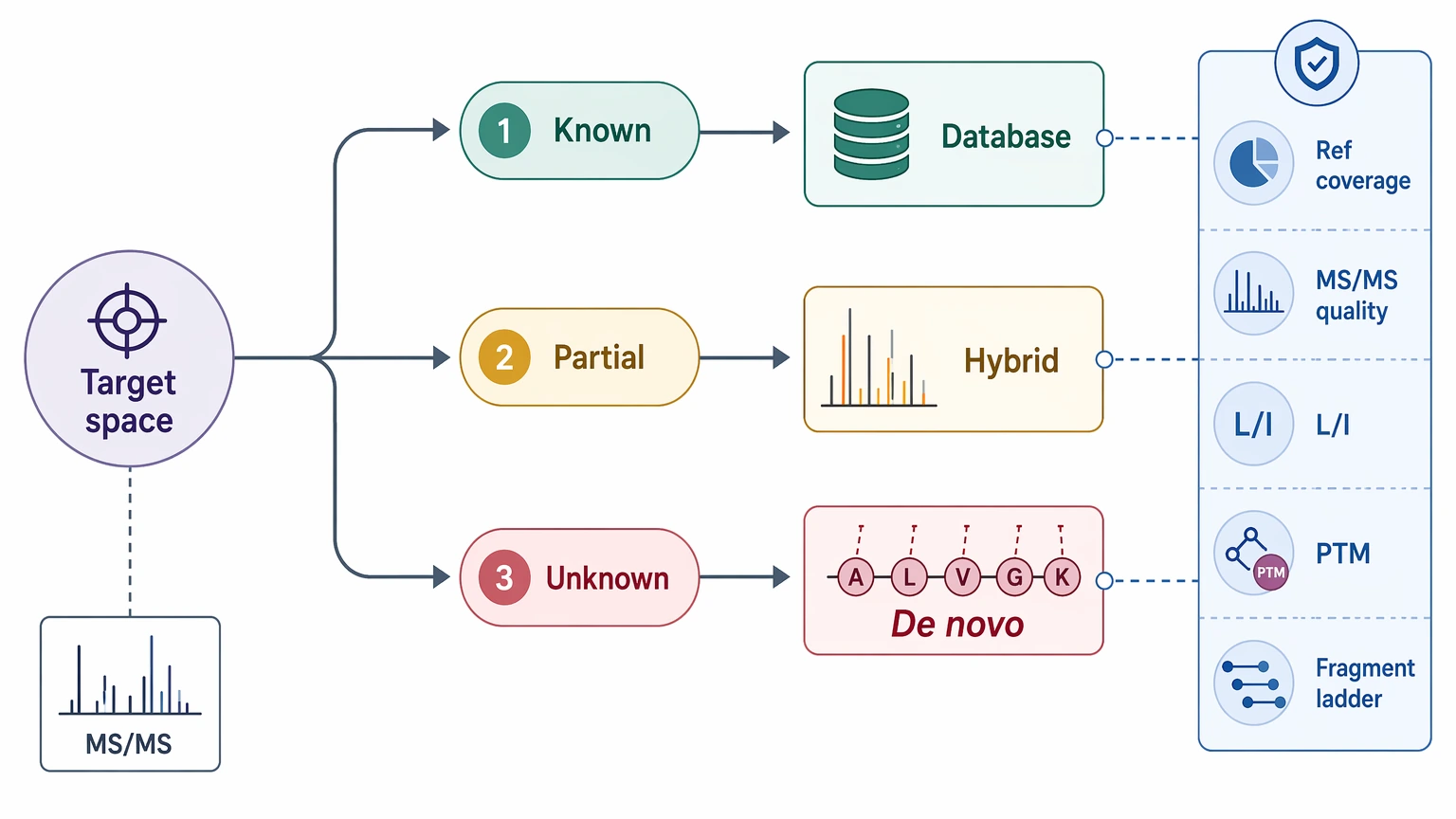
That framing also helps with resource planning. Teams can decide earlier whether the likely output is a final peptide call, a partial sequence tag, a candidate family, or a recommendation to repurify the sample and reacquire data. Those are different outcomes, but each can still support a project decision when it matches the original goal.
MS-based de novo sequencing is most useful in projects involving genuine sequence novelty, incomplete references, unexpected processing, or PTM-rich analytes with interpretable spectra. For peptide discovery, engineered sequence follow-up, or database-limited characterization, the method is strongest when paired with a realistic validation plan and a clear view of what MS/MS alone can and cannot resolve. If your team is weighing de novo sequencing against a database-first or hybrid path, contact us to discuss spectra, sample constraints, and validation goals with MtoZ Biolabs before finalizing the study design.
FAQ
Can de novo sequencing still help if the peptide is too long for full reconstruction?
Yes. Longer peptides often fragment unevenly, so a complete sequence may not be realistic. Even then, shorter sequence tags can still support family-level assignment, custom database building, or targeted follow-up experiments.
Why do some strong MS/MS spectra still produce ambiguous residue calls?
Because some ambiguities come from chemistry, not just noise. Leucine and isoleucine are the classic example, and near-isobaric residue combinations or modification-driven mass shifts can leave more than one plausible explanation.
Is manual spectrum interpretation still useful when software scoring is available?
Yes. Manual spectrum interpretation is often helpful when the highest-ranked candidates are close in score, when PTM localization is uncertain, or when mixed spectra make automated calls look cleaner than they are.
Should teams acquire multiple fragmentation modes for the same target?
Often yes, if sample amount allows it. Complementary CID, HCD, or ETD data can improve sequence continuity or clarify PTM behavior, especially when one fragmentation mode leaves gaps.
What is a good outcome if a project does not return a final sequence?
A useful partial outcome might be a ranked candidate list, a conserved motif, a terminal sequence tag, or a clear explanation that current purity or spectral complexity prevents higher confidence. That can still point the next experiment in the right direction.
When is top-down proteomics more informative than bottom-up de novo sequencing?
Top-down proteomics becomes more informative when intact proteoform context, combinatorial PTMs, or protein-level heterogeneity matters more than reconstructing one bottom-up peptide at a time.
How to order?

