





De Novo Protein Sequencing Service: How to Evaluate Sample Fit, Deliverables, and Vendor Readiness
- the target is unknown, novel, poorly annotated, or only partly represented in databases
- de novo peptide sequencing can add sequence tags beyond a database search
- your team needs peptide assembly evidence, not just an identification label
- you can accept confidence annotation, unresolved regions, and follow-up confirmation where needed
- sample purity is low
- PTM burden or heterogeneity is high
- sample amount does not support repeats or orthogonal validation
- the business question could be answered faster by peptide mapping, intact mass, or standard protein identification
- How do you combine database search with de novo peptide sequencing, and when does each part add value?
- What LC-MS/MS and fragmentation approach is used to support sequence tag generation and fragment ion interpretation?
- When one enzyme gives weak sequence coverage, how do you adjust proteolytic digestion strategy?
- How are confidence score or confidence annotation criteria reported at peptide and assembly levels?
- How do you flag ambiguous residue assignment, isobaric residues, and the leucine/isoleucine limitation?
- When do you recommend intact mass, peptide mapping, top-down support, or other orthogonal validation instead of forcing a complete sequence call?
A de novo protein sequencing service usually makes sense when database search cannot resolve the main project question, the sample can generate interpretable LC-MS/MS data, and the vendor is prepared to show confidence limits instead of presenting one absolute sequence claim. Strong candidates include purified proteins, enriched bands, and focused sequence confirmation projects. Weak candidates include highly mixed, heavily formulated, low-amount, or severely degraded materials unless the goal is narrow and the reporting expectations stay realistic.
Quick decision block
Choose a de novo protein sequencing service when:
Pause or narrow scope when:
Why this outsourcing decision becomes difficult
Most teams do not look into de novo protein sequencing out of general curiosity. They get there because a specific gap opens up: a recombinant product no longer matches the expected record, an archived material lacks a dependable reference sequence, a PTM-rich protein gives incomplete database-search results, or a sequence confirmation task cannot be closed with peptide mapping alone.
That is where the decision often slows down. First, the sample may be available, but no one is sure whether its purity, amount, and heterogeneity are workable for LC-MS/MS-based interpretation. Second, vendor proposals often name the method without explaining the raw data deliverable, annotated spectra, sequence tag evidence, or peptide assembly logic. Third, internal reviewers may see the word “sequencing” across several proposals without a clear way to compare confidence annotation, ambiguous residue assignment handling, or orthogonal validation planning.
For buyers, that is the real issue. The question is not simply whether tandem mass spectrometry can generate useful data. It is whether the project is scoped tightly enough to produce sequence evidence that will hold up under review.
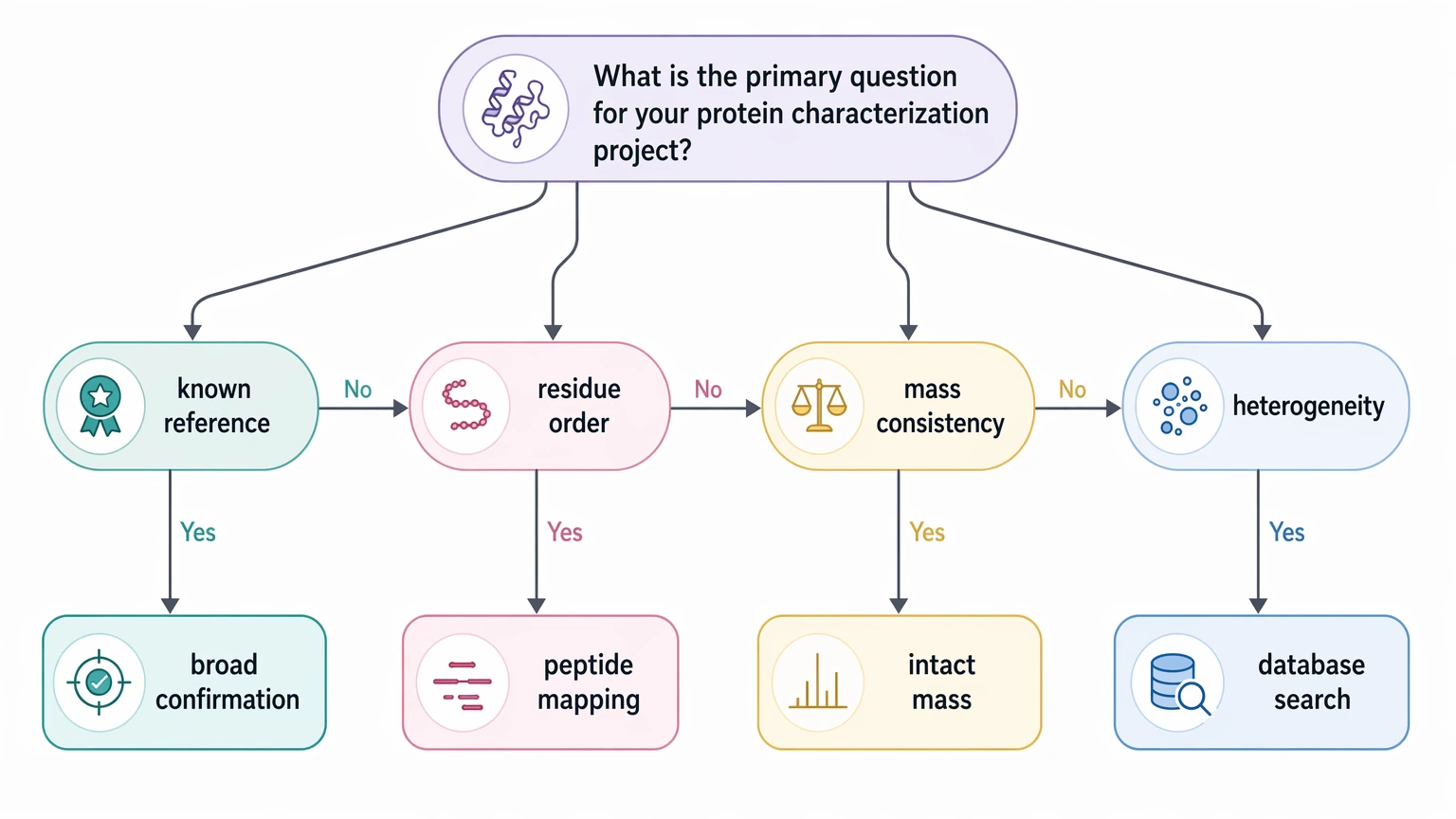
When de novo sequencing is a better fit than database search alone
Database search and de novo peptide sequencing work best as complementary tools. A database search is effective when a close reference already exists and the main job is to match observed peptides to known entries. De novo protein sequencing becomes more relevant when the target sequence is unknown, contains novel regions, has weak database representation, or carries modifications that make straightforward matching less reliable.
A simple rule helps here: if your decision depends on local sequence evidence rather than a best-match identification, de novo interpretation deserves real consideration. That includes unknown protein identification from enriched material, sequence confirmation after unexpected analytical findings, and characterization of proteins with truncation, clipping, or modification patterns that interrupt standard matching logic.
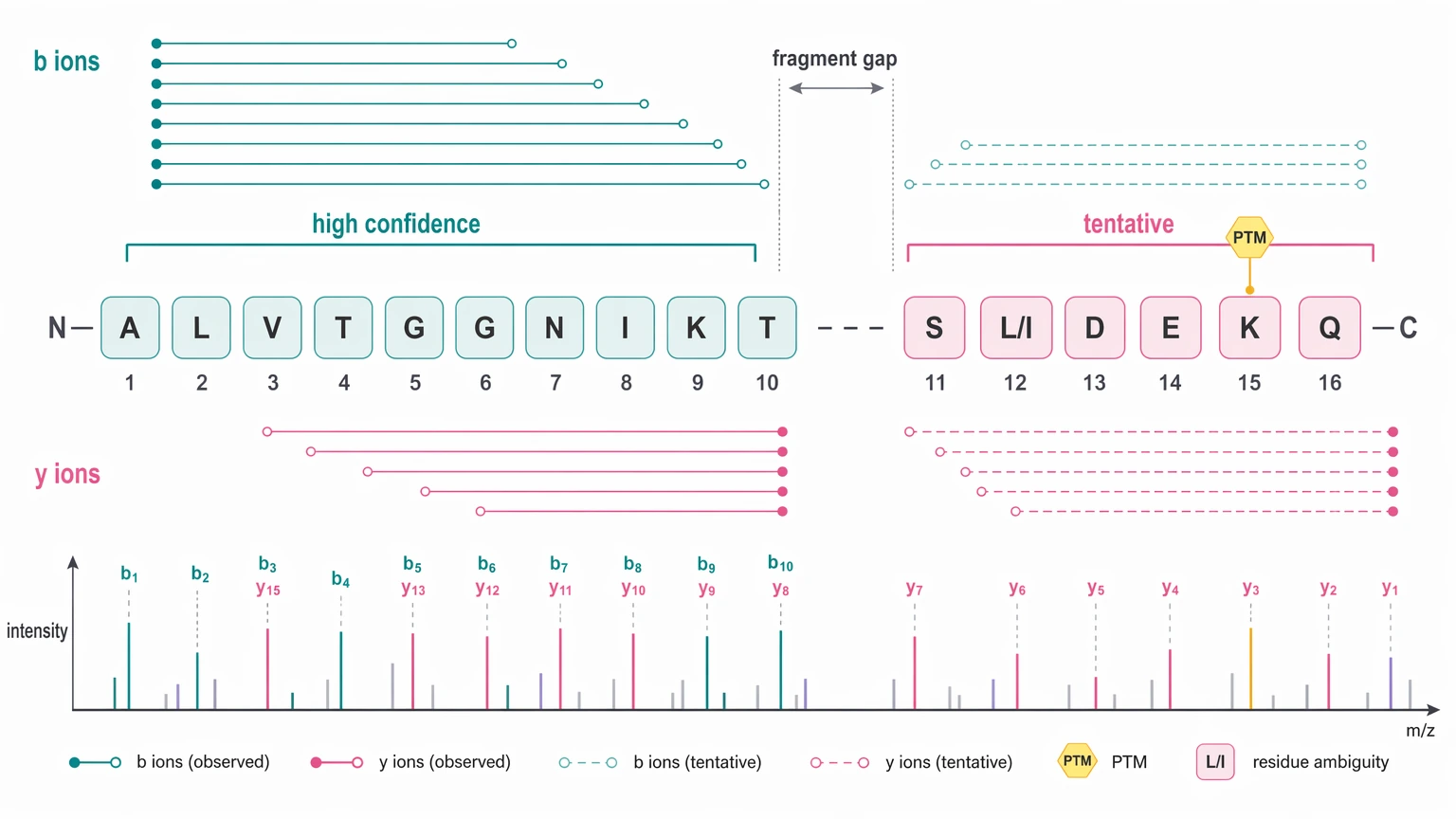
Even then, certainty is not evenly distributed across the sequence. One region may show a strong fragment ion series and consistent b ions / y ions, supporting a high-confidence assignment, while another remains tentative. LC-MS/MS can support strong sequence proposals, but it does not automatically resolve every ambiguous residue assignment. The leucine/isoleucine limitation is the routine example, and PTMs or incomplete fragmentation can add more uncertainty.
Step 1: Judge sample fit before you discuss timing or price
For this topic, four fit categories matter most.
Sample purity and background
The cleaner the target, the easier it is to read fragment ion patterns. Formulation excipients, surfactants, salts, host-cell proteins, and co-migrating bands can reduce spectrum clarity and weaken sequence tag quality. A gel band or enriched fraction may still be workable, but the vendor should explain how contamination risk will be reviewed before final interpretation.
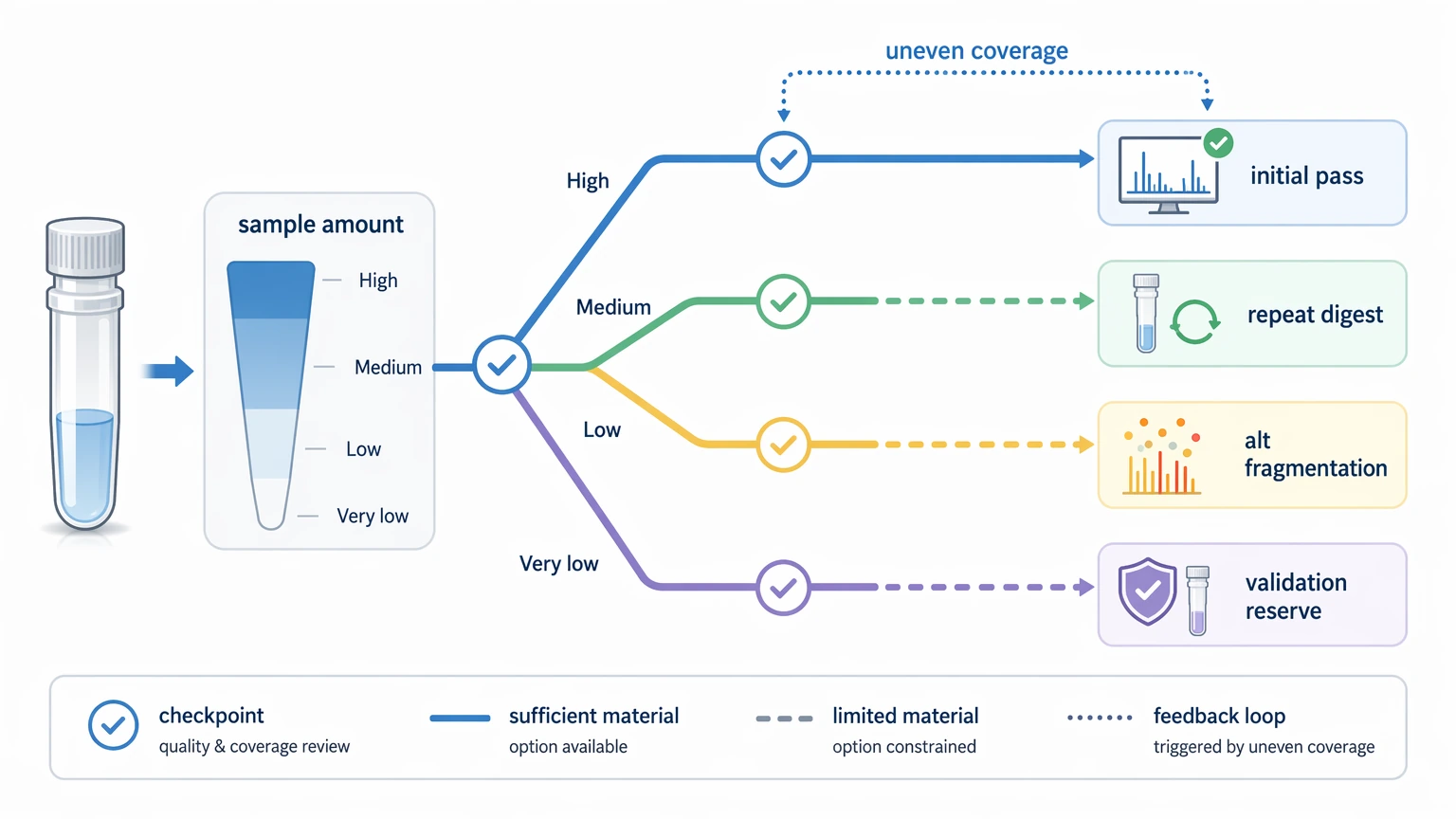
Sample amount and repeat capacity
A small amount may support one bottom-up workflow and nothing more. That leaves no material for repeat proteolytic digestion, alternate fragmentation, or follow-up orthogonal validation. Buyers should ask whether the available amount covers only an initial pass or also leaves room for confirmation work if sequence coverage comes back uneven.
Heterogeneity and PTM burden
Multiple proteoforms, glycosylation, disulfide linkages, truncations, and clipped termini make interpretation harder. These features do not rule out de novo protein sequencing, but they often call for a more deliberate bottom-up workflow, possibly more than one proteolytic digestion strategy, and tighter reporting language. If PTMs are suspected, ask how they will be separated from sequence variation during peptide assembly. One limitation should be stated plainly: MS/MS interpretation may not fully separate sequence novelty from modification effects when fragmentation is incomplete or database representation is weak.
Goal alignment
Unknown protein identification, sequence confirmation, and full-sequence reconstruction are related goals, but they are not the same task. A sample that is suitable for confirming one suspicious region may still be a poor candidate for residue-by-residue claims across the full protein. Buyers should define the business decision first, then ask whether the dataset needs full assembly or only targeted confirmation.
Step 2: Define the report you are actually buying
A credible deliverable should show how the answer was built, not just state the answer. The table below is a practical screen for report quality.
The most useful reporting package usually includes the following items:
| Evidence | What it supports | Main limitation | What to ask next |
|---|---|---|---|
| Raw data deliverable | Independent re-review and reprocessing | Raw files alone do not explain interpretation choices | Ask for acquisition and processing summary |
| Annotated spectra | Residue-level support from fragment ion series | Some spectra will still contain gaps | Review b ions / y ions evidence in hard regions |
| Peptide list with confidence annotation | Strength of de novo peptide sequencing calls | Strong peptides do not guarantee full-protein certainty | Check whether confident peptides cover critical regions |
| Sequence tag summary | Local evidence where database search is weak | Tags may not assemble uniquely | Ask how sequence tags were linked into peptide assembly |
| Assembled sequence proposal | Working protein-level hypothesis | Unresolved positions may remain | Require explicit ambiguous residue assignment notes |
| PTM and termini notes | Boundaries of interpretation | PTMs can mask sequence evidence | Plan orthogonal validation where business risk is high |
A report should also explain whether database search contributed supporting context, where sequence coverage is uneven, and which regions remain provisional. High sequence coverage is useful, but it is not the same as full sequence confidence.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
If your team is already comparing vendors, this is also the point to submit your requirements and evaluate your project against the expected report depth before authorizing sample use.
Step 3: Screen vendor readiness with de novo-specific questions
A vendor is ready for this kind of work when it can explain the logic behind the workflow, not just name the platform. The most useful screening questions are:
A good vendor should also tell you what to prepare before shipment: expected molecular weight, formulation components, known sequence fragments, prior LC-MS/MS files, purity context, and the exact decision the report must support.
When the project involves unknown proteins, PTM-rich material, or database-limited interpretation, a consultation is worth using as a scoping step rather than treating it like a sales formality. Teams can contact MtoZ Biolabs to submit your requirements and evaluate your project against sample format, existing data, and the level of sequence confirmation needed for review.
Expected results and validation methods
A strong project should produce interpretable evidence, not artificial certainty. In the immediate deliverables, expect a method summary, raw data deliverable, annotated spectra, a peptide list, sequence coverage by region, peptide assembly logic, PTM notes, and confidence annotation that separates strong assignments from unresolved or lower-confidence regions.
Follow-up confirmation sits on a different layer. If the proposed sequence will affect comparability conclusions, product understanding, or legacy-material decisions, ask in advance which findings need orthogonal validation. That may include intact mass consistency checks, peptide mapping against the proposed sequence, targeted confirmation of uncertain peptides, top-down support for selected regions, or terminal analysis when N- or C-terminal assignments remain unclear.
The distinction matters. Immediate deliverables show what the LC-MS/MS dataset supports now. Follow-up confirmation tests whether uncertain regions, termini, or PTM-related interpretations still stand when checked with a second method.
Key cautions and practical limits
Several limits should stay in view before budget approval.
Low sample quality or low sample amount can restrict repeat analysis, alternate digestion, and confirmation work. If the available material supports only one attempt, the reporting scope should be narrower.
Controls and repeats should match the business question. For sequence confirmation, preserve comparison material when possible. For unknown protein identification from enriched but mixed samples, ask how repeat expectations change if contamination or co-eluting species are detected.
Batch effects and contamination risk matter when material comes from multiple lots, gels, or archived preparations. Document source, storage history, formulation, and cleanup steps before shipment so unexpected peptides are interpreted in context.
Interpretation boundaries matter too. LC-MS/MS-based de novo protein sequencing can support strong local assignments, but some residue positions may remain uncertain because of incomplete fragmentation, isobaric residues, PTMs, or database-search limits. A responsible report should mark those boundaries clearly.
Sometimes another method is simply the better next step. If the question is standard protein identification in a known reference space, a database search-focused workflow may be faster. If the main uncertainty is mass consistency, truncation, or heterogeneity rather than residue order, intact mass or peptide mapping may answer the decision more directly. If internal review will require broad orthogonal confirmation, outside support should be planned from the start rather than added after a borderline report.
Conclusion
A de novo protein sequencing service is the right fit when your project needs sequence evidence beyond a database search, your sample can support interpretable LC-MS/MS data, and the vendor is prepared to document confidence limits, unresolved regions, and validation options. That framework is especially useful for unknown protein identification, sequence confirmation after unexpected findings, and database-limited or PTM-rich samples where peptide assembly needs careful interpretation. If your team is deciding between de novo sequencing, peptide mapping, intact mass, or a combined workflow, contact MtoZ Biolabs to discuss the sample context, technical limits, and the most defensible reporting plan before committing material and budget.
FAQ
Can a vendor work from my existing LC-MS/MS files instead of receiving new samples?
Sometimes, yes. Existing files can support an early feasibility review, especially when the question is whether de novo peptide sequencing is likely to add value. Still, the vendor may recommend new acquisition if fragmentation quality, precursor selection, or digestion design limits sequence confidence.
What is the clearest sign that a proposal is overstating certainty?
Watch for proposals that promise a final full sequence but do not mention annotated spectra, confidence annotation, unresolved regions, or orthogonal validation. In de novo work, silence about uncertainty is usually a stronger warning sign than a modest sequence-coverage estimate.
Should I ask for one enzyme or multiple digestion conditions?
Ask how the vendor decides. A single proteolytic digestion may be enough for a simple confirmation task, but heterogeneous or modification-rich proteins often benefit from a multi-enzyme approach to improve peptide distribution and sequence coverage.
How should I package project background for a feasibility review?
Provide the target type, expected molecular weight, known sequence fragments, suspected PTMs, formulation components, purity data, gel images if available, prior tandem mass spectrometry results, and the exact decision the report must support. That shortens the technical review and reduces scope mismatch.
Is top-down support mandatory for de novo protein sequencing?
No. Many projects are built around a bottom-up workflow. Top-down support becomes more useful when proteoform structure, termini, or intact-region confirmation matters and the sample quality can support that extra layer.
What if my main goal is to compare a suspect sample with a known reference?
That may still justify de novo work, but the scope should usually focus on sequence confirmation in the regions of concern. If the reference is strong and the uncertainty is narrow, peptide mapping plus targeted confirmation may be more efficient than a broad de novo assembly project.
How to order?

