





De Novo Assembly Shotgun Sequencing: What Affects Contig Quality, Repeat Resolution, and Assembly Usability
- Sample quality or amount limits: Low abundance, degradation, salt carryover, or poor enrichment can reduce readable spectra before assembly starts.
- Controls and repeat expectations: Technical replicates or complementary runs often matter because repeated overlap can separate a stable consensus sequence from a chance local call.
- Batch or contamination risk: Mixed backgrounds, keratin, carryover, or co-purified related proteins can create misleading continuity and increase false sequence path risk.
- Interpretation boundaries: A strong contig does not by itself confirm full protein identity, complete PTM architecture, or exact residue assignment at every ambiguous site.
- When another method is better: If the key question is a critical leucine/isoleucine position, exact terminal assignment, or a heavily modified repeat region, a complementary sequencing method or outside support may be the more efficient next step.
De novo assembly shotgun sequencing is most useful when shotgun LC-MS/MS produces de novo peptide sequencing tags that overlap cleanly, hold local confidence across adjacent residues, and point back to one protein backbone instead of several competing explanations. It becomes much less usable when fragment-ion ladders are incomplete, overlap depth is thin, homologous proteins are mixed together, or post-translational modification creates more than one credible sequence path. For most teams, the first practical question is not whether de novo protein sequencing can be attempted. It is whether the current data can yield a contig that is specific enough to support the next project decision.
A usable output does not always mean a full-length consensus sequence. In many projects, partial success is enough. A contig may support protein family assignment, anchor a distinctive motif, define a region for orthogonal validation, or show that a database search failed because the sample contains a novel segment rather than simple low quality. Standard tandem mass spectrometry still has interpretation limits, though, and some positions remain uncertain even in otherwise strong data sets.
Quick Decision Guide
| If your data show... | Most likely interpretation | Best next step |
|---|---|---|
| Long sequence tags with repeated overlap in the same region | Contig extension is technically plausible | Continue overlap assembly and map usable regions |
| Good peptide calls but little bridging overlap | Digestion geometry is limiting assembly | Revisit protease specificity or add complementary digestion |
| Strong evidence plus mixed or related proteins | Ambiguity is biological, not only computational | Reduce complexity or validate distinguishing regions |
| PTM-rich peptides with branching interpretations | Backbone and modification states are competing | Separate core sequence questions from PTM localization follow-up |
Takeaway: judge assembly usability against the downstream task, not against an all-or-nothing full-sequence expectation.
What “Assembly” Means in Shotgun LC-MS/MS
In this context, assembly is not genome-style reconstruction. It is overlap assembly of de novo peptide sequencing outputs derived from shotgun LC-MS/MS. Each MS/MS spectrum supports a local sequence call through fragment ions, often dominated by b ions and y ions, and sometimes complemented by c ions and z ions depending on fragmentation mode. Those local calls become sequence tags. A longer contig forms only when multiple tags overlap in a consistent way and support one consensus sequence more strongly than alternative paths.
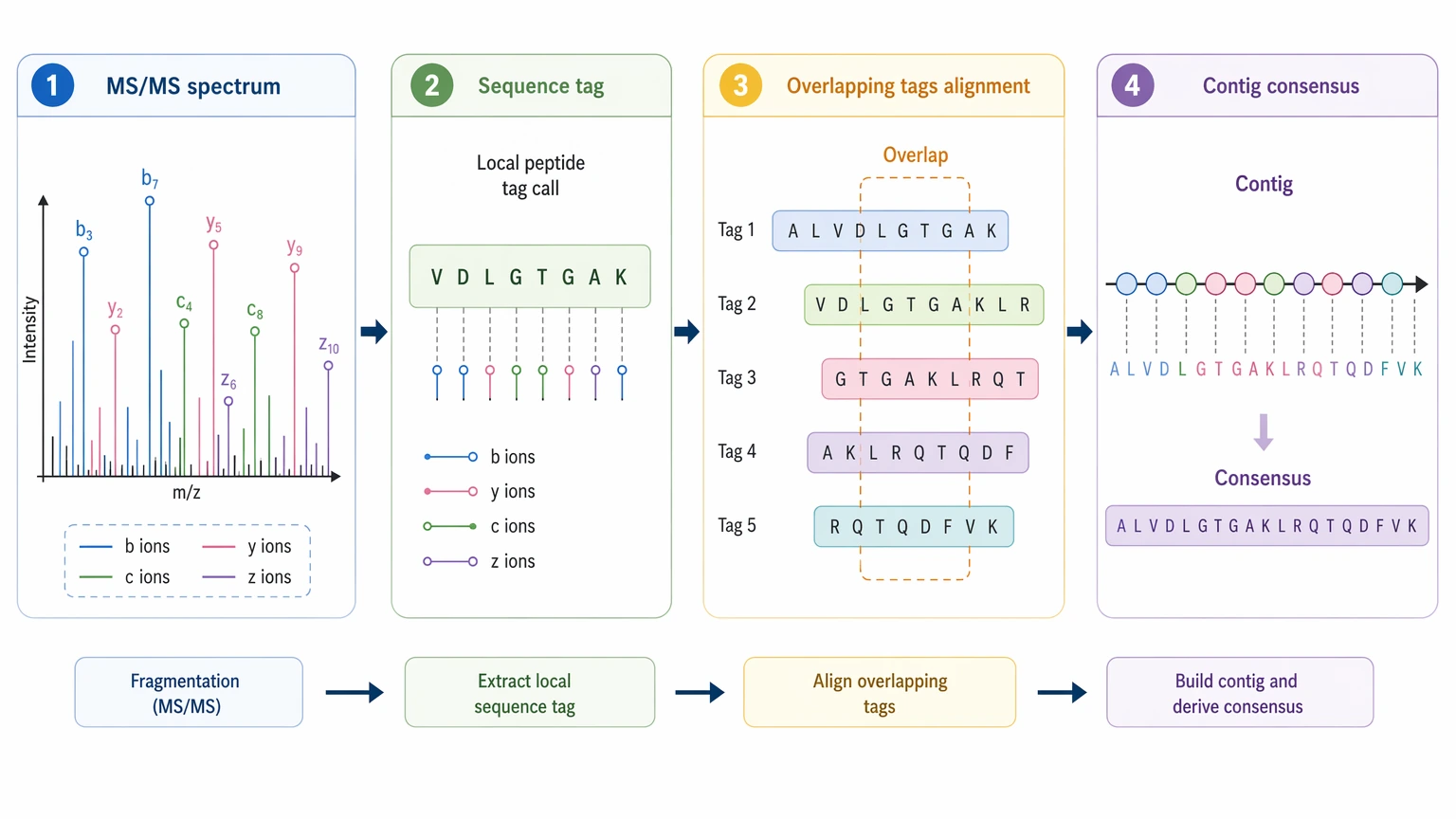
That distinction matters because a peptide-spectrum match and a de novo sequence call answer different questions. A peptide-spectrum match tests whether a known sequence fits the data. De novo peptide sequencing infers residue order directly from the spectrum. De novo protein sequencing extends that inference by linking peptides into a broader protein-level interpretation. If database searching is weak because the target is novel, engineered, or poorly annotated, assembly-like reconstruction may still recover useful evidence. It can also fail for reasons that have very little to do with software choice.
The Main Drivers of Contig Quality
Four factors usually decide whether contigs stay short, extend cleanly, or break into conflicts.
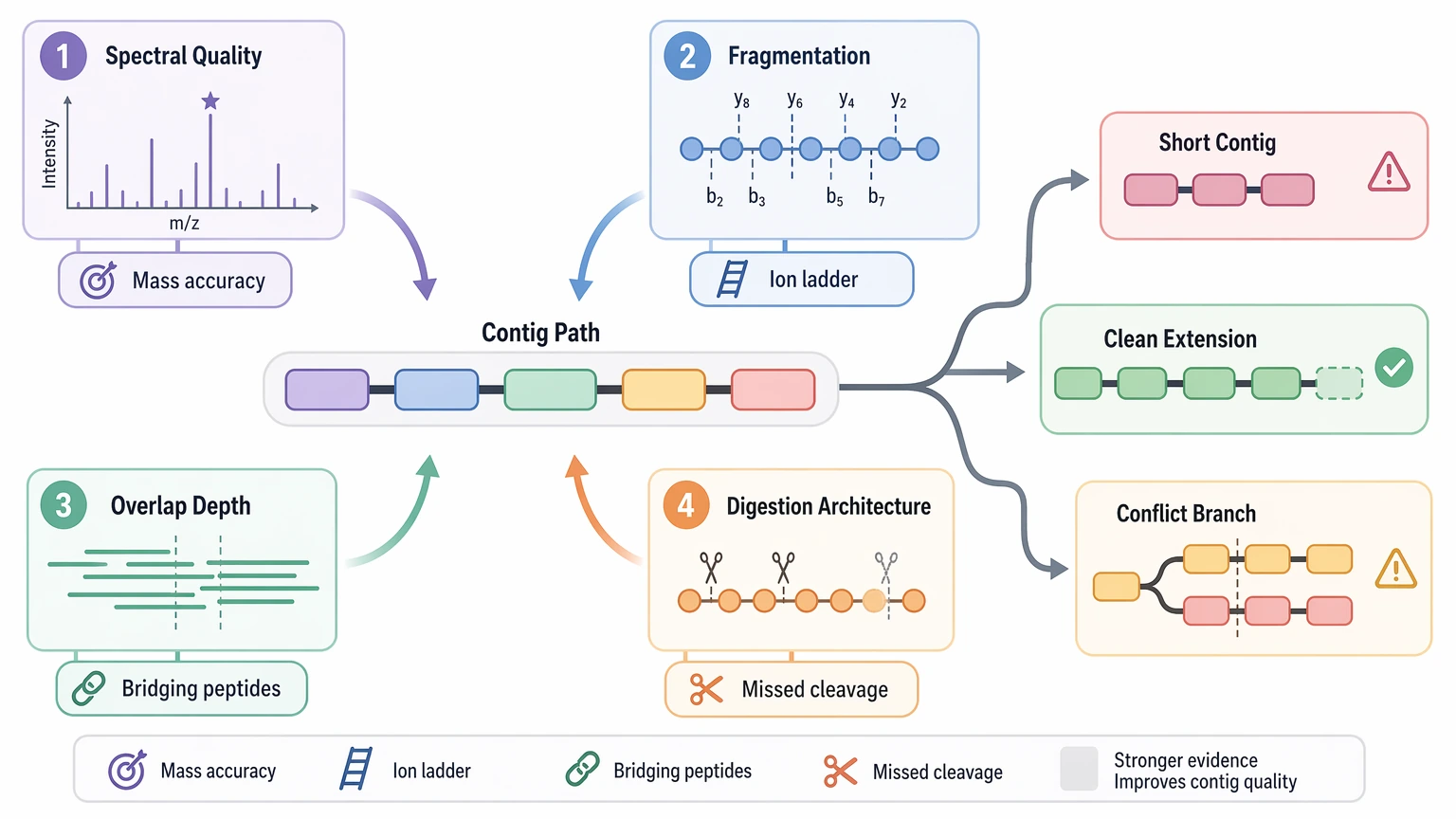
Spectral quality and mass accuracy
De novo assembly starts with local spectral interpretability. Strong precursor mass accuracy, strong fragment mass accuracy, readable ion ladders, and limited co-isolation make residue-to-residue inference more credible. If the MS/MS spectrum is noisy or dominated by a chimeric spectrum, confidence drops before overlap assembly even starts.
Fragmentation behavior
A peptide that fragments into a continuous ion series contributes much more than one with only a few isolated peaks. CID, HCD, ETD, or hybrid strategies can change how much sequence information is visible and whether labile modifications are retained. No single fragmentation mode is always best. The more useful question is whether the chosen mode exposes enough complementary ion evidence for this peptide population.
Overlap depth
A single high-confidence tag is informative, but a stable contig usually needs multiple overlapping peptides across the same region. Overlap depth determines whether one uncertain local call is corrected by neighboring evidence or remains a break point. When overlap is shallow, contigs often look longer than they really are because local tags cannot be checked against redundant support.
Digestion architecture
Protease specificity, peptide length distribution, and missed cleavage burden shape the geometry of assembly. Very short peptides carry limited information. Very long peptides may fragment unevenly. Uneven digestion can leave good local evidence in place but still miss the bridging peptides needed to connect adjacent regions.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
Before changing software settings, use the pattern below to identify the real bottleneck.
| Evidence pattern | What it usually means | Most useful response |
|---|---|---|
| Short tags and weak ion ladders | Local sequence confidence is low | Improve acquisition quality or fragmentation fit |
| Many peptides but few overlaps | Digestion produced isolated tags | Redesign digestion for better bridging |
| Strong tags in only part of the protein | Chemistry is region-dependent | Evaluate charge state and peptide properties |
| Abundant spectra from a mixed sample | Signal is present but not uniquely assignable | Simplify the sample or narrow the target |
Takeaway: contig failure often starts upstream of assembly scoring.
Why Repeat Resolution Breaks Down
Repeat resolution in de novo protein work usually means separating similar local explanations, not assembling long DNA repeats. Low-complexity motifs produce low-information tags. Conserved domains from homologous proteins can overlap so strongly that multiple placements stay plausible. Isoform ambiguity creates the same problem when peptides fit more than one related backbone.
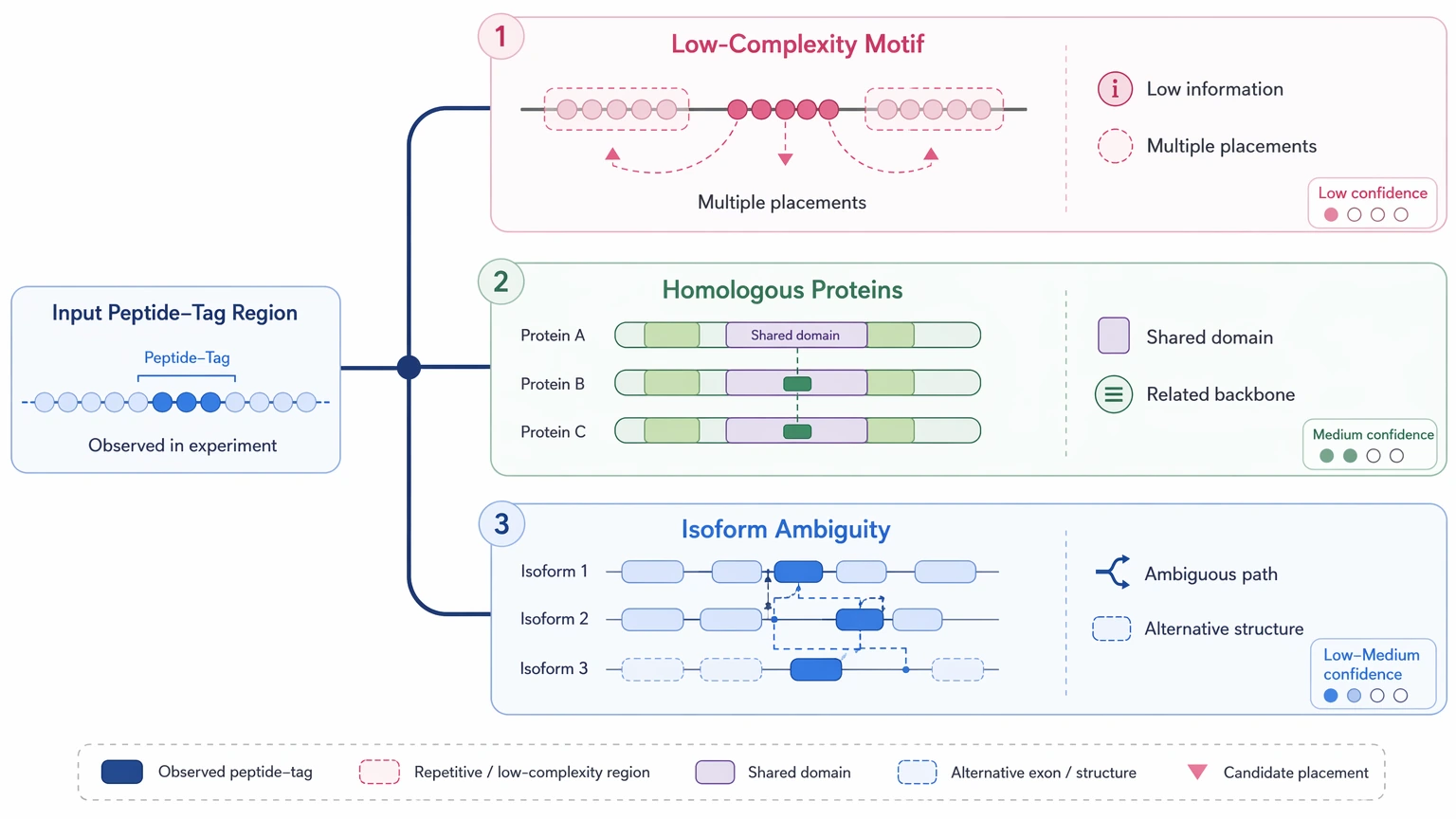
Two additional issues are especially persistent. First, isobaric residues create unresolved identity at specific sites, with leucine/isoleucine ambiguity as the classic example. Second, a post-translational modification can mimic part of the mass shift expected from a variant, especially when PTM localization is incomplete. In those regions, more than one sequence path may fit the observed fragments.
An explicit limitation matters here: standard shotgun LC-MS/MS often cannot prove a single unique residue path across all ambiguous positions, especially in PTM-rich or repeat-rich regions. De novo outputs should therefore be treated as confidence-graded evidence, not automatic proof of one final full-length sequence.
When a Partial Contig Is Still Usable
Assembly usability should be tied to the real decision in front of the team. A partial contig may already be useful when the goal is to assign a family, localize a motif, identify a novel insertion, design targeted follow-up, or refine a custom database for a second search round. In those settings, sequence coverage can be incomplete while the output is still actionable.
Usability drops when the next step requires exact residue identity across ambiguous positions, unique discrimination among near-identical family members, or complete modification mapping. For clone design, regulatory documentation, or hard novelty claims, an incomplete contig may be informative but not sufficient.
If your data already contain credible local tags yet the project turns on whether those tags can support synthesis planning or targeted confirmation, you can contact MtoZ Biolabs to evaluate your project against the actual overlap pattern, expected uncertainty, and intended follow-up rather than against a generic expectation of full-length recovery.
Expected Results and How to Validate Them
A realistic de novo assembly project should separate immediate analytical deliverables from later confirmation.
Immediate deliverables
The first deliverable is usually a ranked set of peptide-level sequence tags, contigs, confidence profiles, and conflict annotations. Useful outputs may include region-specific sequence coverage, overlap maps, unresolved positions, PTM-aware candidate paths, and notes on whether ambiguity comes from poor spectra, mixed proteins, or true structural similarity.
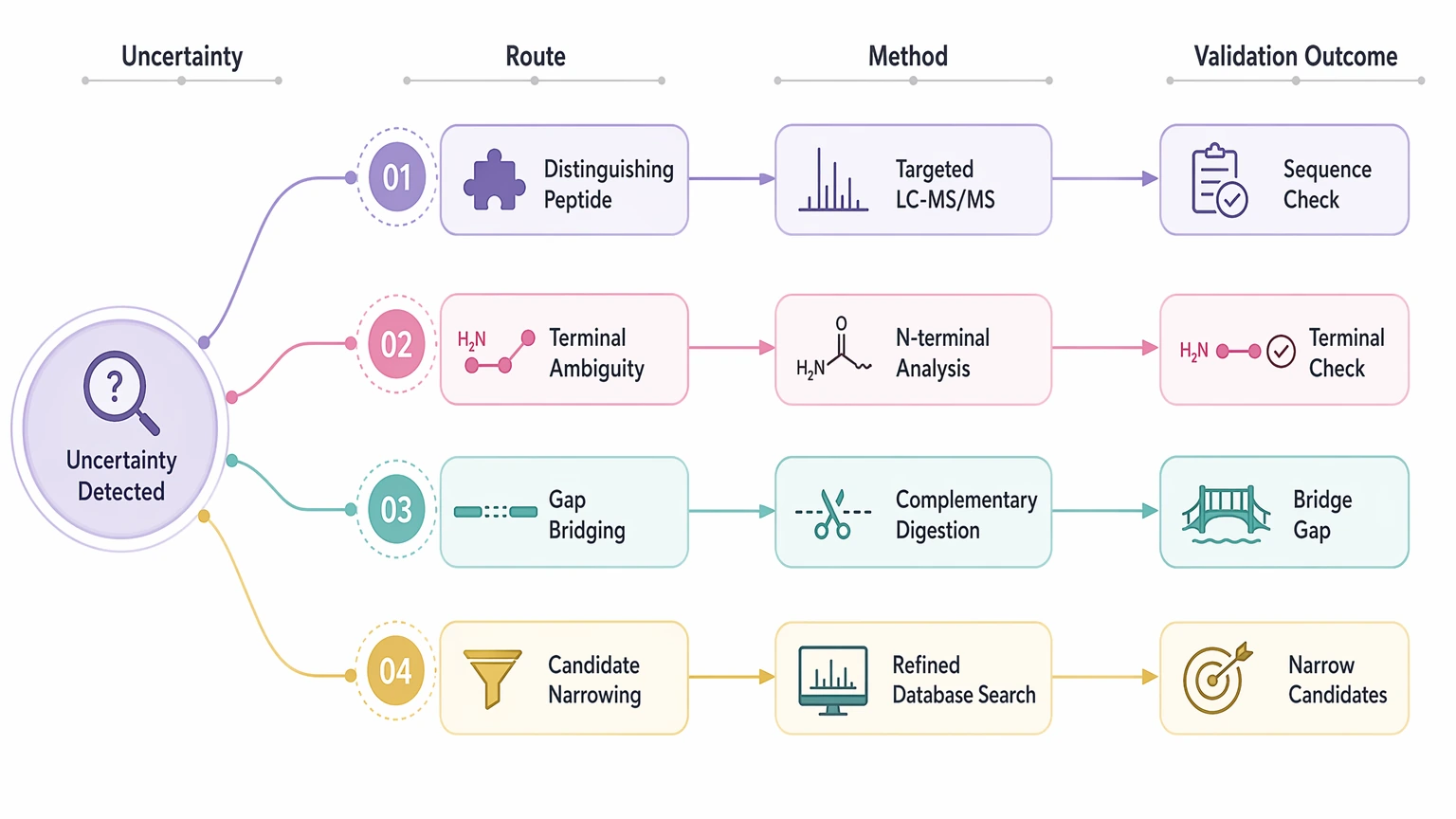
Follow-up confirmation
Confirmation comes later and should target the exact uncertainty that still matters. Orthogonal validation may include targeted LC-MS/MS for a distinguishing peptide, Edman-style N-terminal work for terminal ambiguity, complementary digestion to bridge a gap, or a refined database search after candidate narrowing. Validation is not only about proving the assembly correct. It is also about testing whether the remaining uncertainty changes the biological or engineering decision.
Key Cautions and Practical Limits
Several practical limits should be stated early so the contig is not overread.
When uncertainty is concentrated in one region rather than spread across the whole protein, the better workflow is often targeted confirmation rather than broad reacquisition. If that is where your team is stuck, submit your requirements to MtoZ Biolabs and evaluate your project using the sample context, acquisition details, and downstream acceptance criteria that actually determine usability.
Conclusion
De novo assembly shotgun sequencing is most informative when shotgun LC-MS/MS produces readable local sequence evidence, enough overlap assembly support to extend beyond isolated tags, and a biological background simple enough to avoid repeated false branching. In unknown proteins, engineered constructs, toxin-like mixtures, or PTM-rich samples, partial contigs can still be useful when they answer a bounded question such as motif localization, candidate narrowing, or validation targeting. If your next step depends on whether current contigs are sufficient for confirmation, redesign, or another acquisition round, prepare the sample history, digestion plan, MS/MS conditions, and decision target before seeking a technical review.
FAQ
Can a strong database-search result still be useful in a de novo project?
Yes. Even when database searching returns plausible matches, de novo interpretation can reveal novel segments, substitutions, or modification-heavy regions that the database result smooths over. It is often most useful as a cross-check, not as a replacement for all search-based analysis.
Are technical replicates worth it for contig building?
Often yes, especially when overlap depth is marginal. Replicates can add repeated evidence for the same region and help separate a reproducible path from a one-off low-confidence call.
Does one missed cleavage always hurt assembly?
Not necessarily. A small number of missed cleavages can sometimes create longer bridging peptides that help connect regions. The problem is heavy or uneven missed cleavage, which leaves gaps and inconsistent peptide geometry.
How should teams report ambiguous residues in internal project decisions?
Use explicit notation for unresolved positions and state why they remain unresolved, such as leucine/isoleucine ambiguity, incomplete fragment coverage, or PTM-versus-variant uncertainty. That makes downstream validation planning much clearer.
When is sample simplification more useful than another acquisition run?
When the spectra are abundant but the contigs keep collapsing into related alternatives, the limiting factor is often mixture complexity rather than instrument sensitivity. In that case, enrichment, fractionation, or isolation of the target component may help more than simply collecting more of the same data.
How to order?

