





Antibody Diversity And Specificity Analysis Troubleshooting: Causes, Checks, and Fixes for Research Workflows
- Antibody Sequencing Service | Mass spectrometry for sequence-resolved review of variable-region evidence and clonotype separation.
- De Novo Peptide Sequencing Services for strong spectra that still resist reference-based matching.
- AAE-nanoLC-MS/MS Antibody Coverage Analysis Service for coverage mapping when the main uncertainty is CDR recovery.
- LC-MS/MS-Based Targeted Site Validation Service for follow-up confirmation of discriminative peptides or modified sites.
Quick Answer
When antibody diversity or specificity analysis gives low-confidence, contradictory, or incomplete findings, check the workflow in this order: sample quality and enrichment bias first, proteolytic digestion second, LC-MS/MS data quality third, and interpretation strategy last. A move to de novo peptide sequencing or de novo protein sequencing is usually justified when unmatched spectra stay concentrated in the variable region, CDR / complementarity-determining region evidence is weak, and the peptide-spectrum match (PSM) rate remains low after sample and acquisition issues have been addressed.
Key limitation: even strong tandem mass spectrometry data does not resolve every antibody sequence question. PTMs, incomplete fragment-ion ladders, database search limitation, and peptide assembly ambiguity can leave some CDR-containing peptides or heavy chain / light chain assignments only partly supported.
Decision Block: Which Workflow Should You Choose Next?
The first routing decision is usually easier when the observed pattern is reduced to one primary failure signal rather than a full list of symptoms.
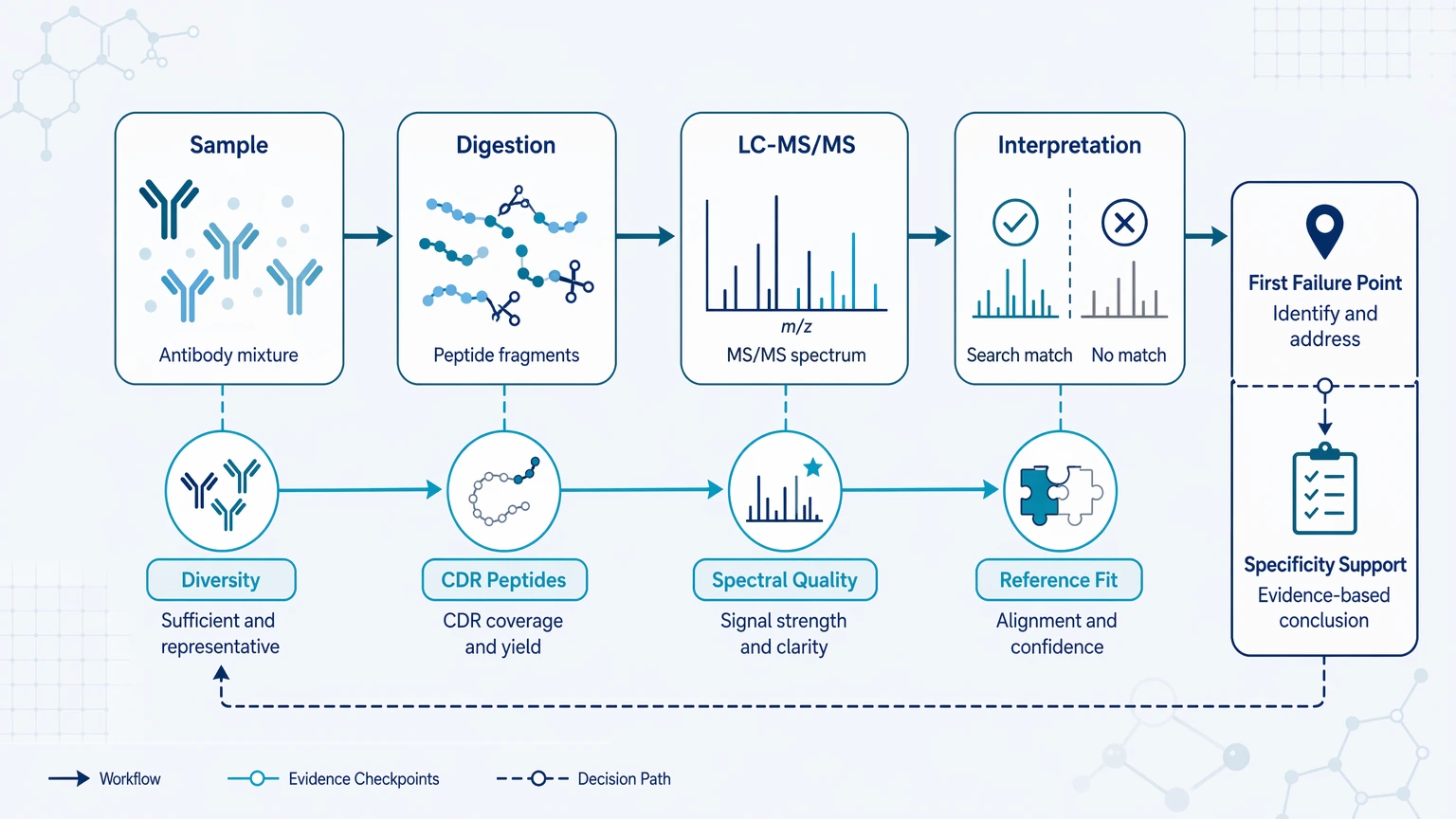
Use the table to connect the dominant failure signal to the first check, then confirm whether the limitation matches the available sample and LC-MS/MS evidence.
| Observed pattern | Check first | Recommended workflow | Key limitation |
|---|---|---|---|
| Apparent diversity drops after purification or enrichment | Sample recovery bias, contaminant background, sample-input sufficiency | Repeat prep or compare with orthogonal material checks | Total peptide count can mask a diversity collapse artifact |
| Strong spectra but many unmatched spectra in variable-region peptides | Reference-sequence incompleteness, precursor isolation quality | Add de novo peptide sequencing to priority spectra | Unmatched spectra do not prove novel antibodies |
| Total coverage looks acceptable but CDR evidence is sparse | Proteolytic digestion design, missed cleavage burden | Redigest with alternate protease and reacquire | Framework-region coverage cannot stand in for CDR evidence |
| Specificity interpretation conflicts with binding data | Shared peptides, PTM burden, chain assignment | Reset sequence claims and use orthogonal validation | Partial sequence evidence can overstate specificity |
| Closely related candidates cannot be separated | Unique peptide count, clonotype discrimination logic | Focus on discriminative variable-region peptides | Redundant peptides inflate confidence without resolving candidates |
This triage works best when the question is sequence-resolved diversity or specificity, not general protein identification.
What Usually Goes Wrong First
The first visible symptom is often not the root cause. A low-diversity readout may reflect real biological enrichment, but it can also come from selective loss of low-abundance antibodies, poor sample-input sufficiency, or reliance on shared framework region peptides that do not support clonotype discrimination. An apparent specificity shift may likewise come from co-isolated peptides, uncertain heavy chain / light chain assignment, or overreading a few incomplete spectra rather than a true change in target recognition.
That is why troubleshooting should follow the evidence chain, not just the protocol. The aim is to find the first failure point that can distort the decision: does the sample contain the expected diversity, and does the sequence evidence support the claimed specificity pattern?
Step 1: Check Sample Quality Before Reinterpreting the Data
Start with the sample. No interpretation method can recover information that was never retained. For antibody mixtures, affinity-enriched fractions, engineered constructs, or partially purified preparations, the main risks are contaminant carryover, selective enrichment bias, degradation, and low input.
Common warning signs include strong constant-region signal with weak variable-region evidence, unstable heavy chain and light chain balance across replicates, high contaminant background from host proteins or affinity reagents, acceptable total ion signal but low unique peptide count, and replicate shifts that mainly affect low-abundance candidates. These patterns can look more complete than they are because many identified peptides may come from conserved regions that do not separate related antibodies.
Checks should focus on whether enrichment matches the decision question, whether discriminative variable-region peptides are preserved, whether handling introduced oxidation or deamidation, and whether the available amount supports sequence-resolved analysis rather than bulk confirmation. Fixes usually involve repeating cleanup only when background is clearly interfering, narrowing the comparison when input is limited, and adding orthogonal material checks such as intact-mass review before making sequence-level claims.
Step 2: Test Whether Proteolytic Digestion Produced the Right Peptides
If the sample is usable, move to proteolytic digestion. Many antibody projects fail here because the peptides that matter most for diversity and specificity are also the hardest to generate and interpret. CDR-containing segments may be too long, too short, modified, or poorly fragmented after a single digestion strategy.
A common failure pattern is good framework-region coverage with poor CDR evidence. That can support broad antibody presence, but not confident comparison between closely related candidates.
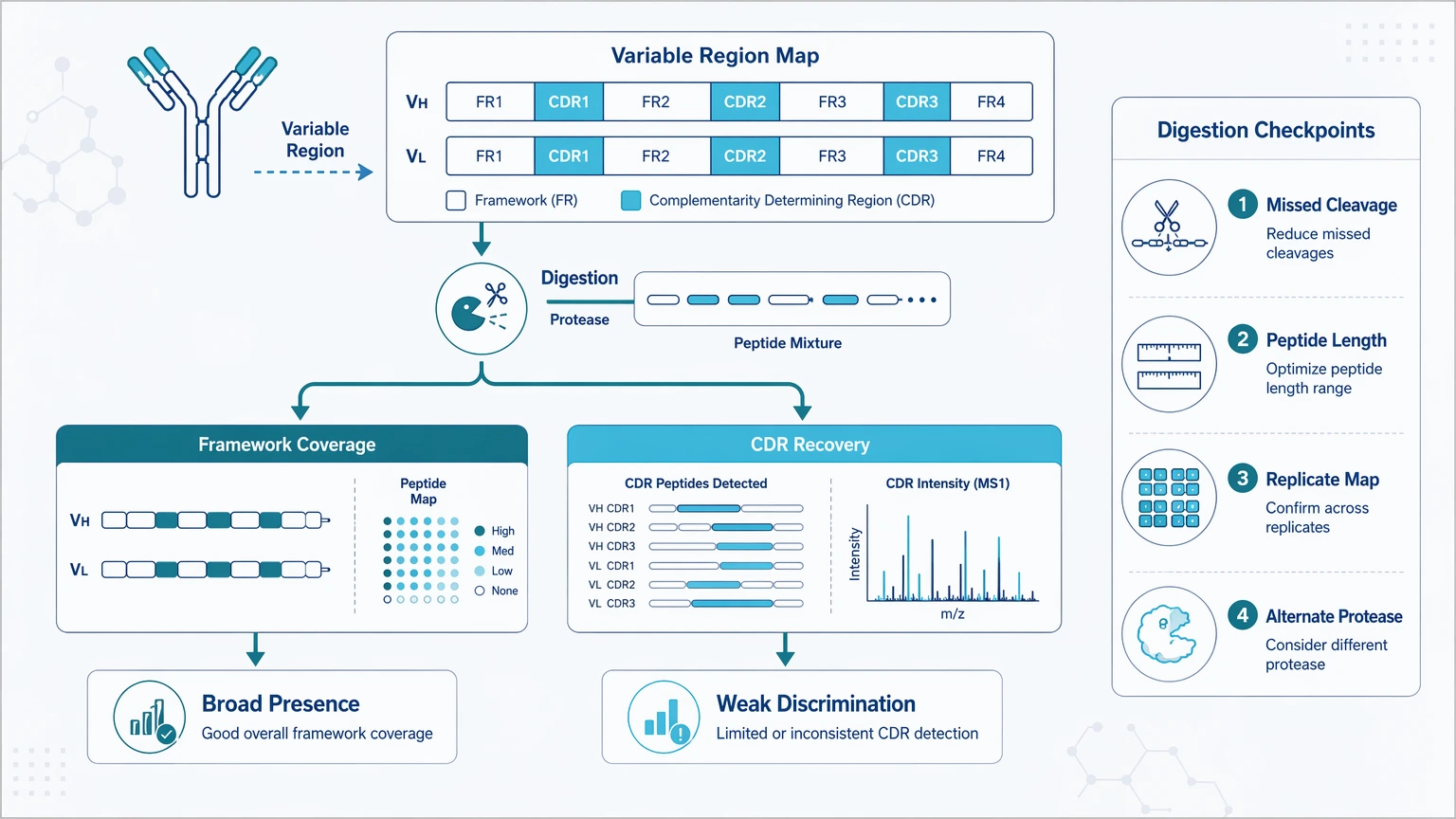
The most useful digestion metrics are sequence coverage in heavy and light chain variable regions, recovery of CDR-containing peptides, missed cleavage burden, peptide length distribution, overlap between enzyme strategies, and consistency across replicate digests. Review where coverage gaps occur, compare peptide maps across technical replicates, and ask whether one enzyme mainly produces redundant framework peptides. When CDR recovery remains weak, an alternate or multi-enzyme plan and targeted reacquisition of priority peptides are usually more informative than rerunning the same digest unchanged.
Step 3: Review LC-MS/MS Depth and Spectral Quality
Once digestion has been checked, evaluate LC-MS/MS performance with antibody-relevant metrics rather than generic identification totals. For diversity and specificity work, the useful question is not how many peptides were identified, but how much interpretable evidence exists in the variable region and how reproducible it is.
Poor acquisition depth can make minor clonotypes disappear, which looks like reduced diversity. Co-isolation can also create mixed fragmentation and conflicting assignments, which lowers sequence confidence and complicates specificity interpretation.
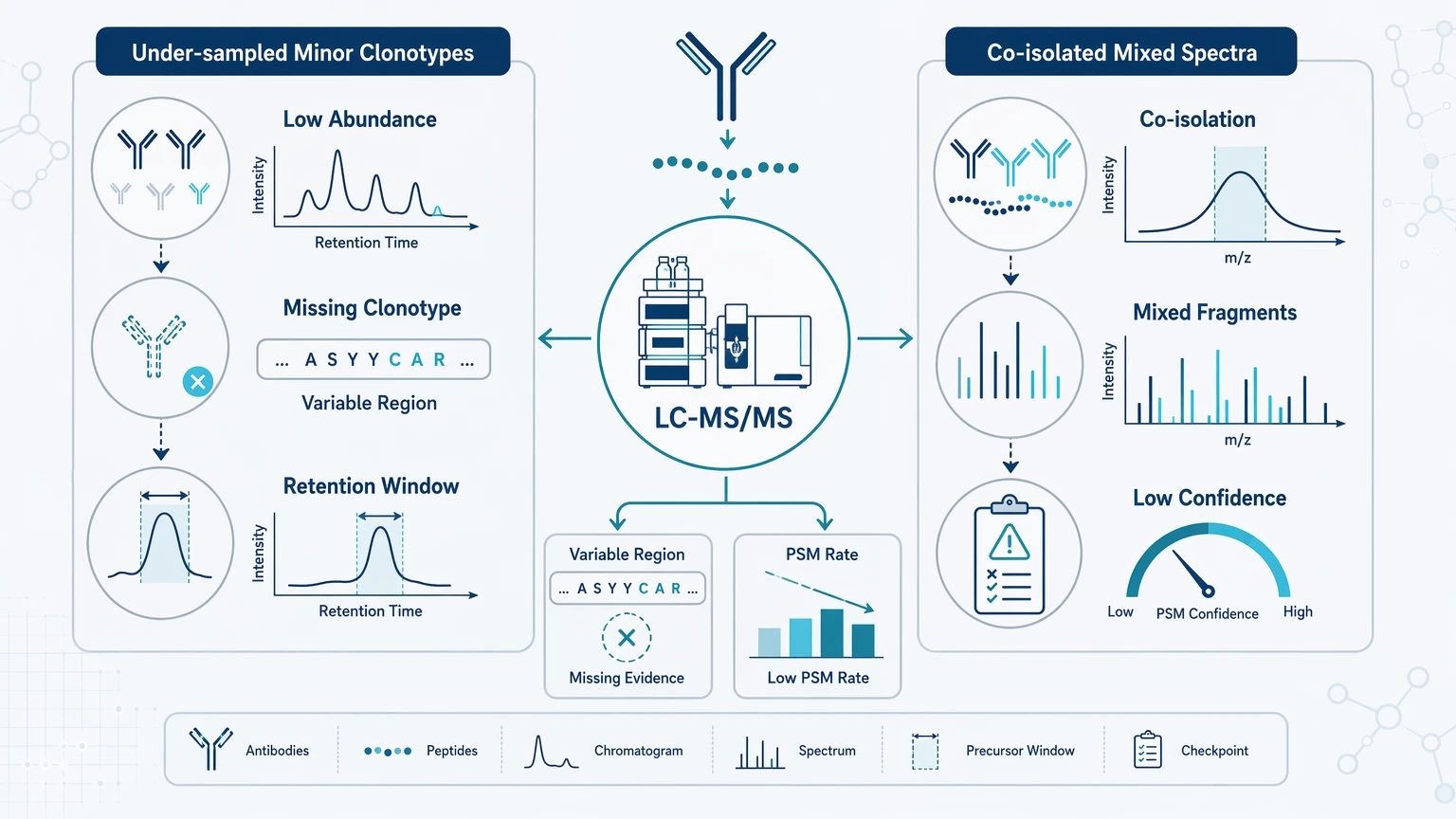
Useful checks include whether unmatched spectra cluster in narrow retention-time windows, how PSM behavior differs between constant, framework, and CDR-containing peptides, and whether longer or lower-abundance variable-region peptides were systematically under-sampled. Fixes may include reacquisition settings that better capture low-abundance or longer peptides, separating discovery runs from targeted confirmation runs, and prioritizing peptides that distinguish candidate antibodies rather than optimizing only total identification count.
If the same dataset keeps failing in the same regions, a project-fit review may save time. Teams deciding whether to escalate from database-only interpretation to sequence-focused troubleshooting can ask MtoZ Biolabs to evaluate your project against an Antibody Sequencing Service | Mass spectrometry or De Novo Peptide Sequencing Services workflow when the sample type, raw LC-MS/MS status, and unresolved variable-region evidence are already defined.
Step 4: Decide Whether the Main Problem Is Reference Mismatch
If the spectra are reasonably strong but the variable-region PSM rate stays low, ask whether the reference model is failing. A conventional database search works best when the antibody sequence is known or closely represented. It becomes much less informative when the sample contains undocumented variants, engineered antibodies, unknown antibodies, or incomplete reference records.
A likely reference-sequence incompleteness pattern includes stable framework-region matches with poor CDR assignments, repeated high-quality unmatched spectra in variable-region peptides, disagreement between search engines on the same spectra, low match recovery despite good mass accuracy, and conflict between database output and manual spectrum review.
This is where de novo peptide sequencing becomes useful. Instead of forcing a reference-dependent match, it derives peptide candidates directly from the MS/MS evidence. De novo protein sequencing becomes relevant when peptide-level findings need to be assembled into a broader characterization question, although the final report should still mark unresolved regions and competing interpretations.
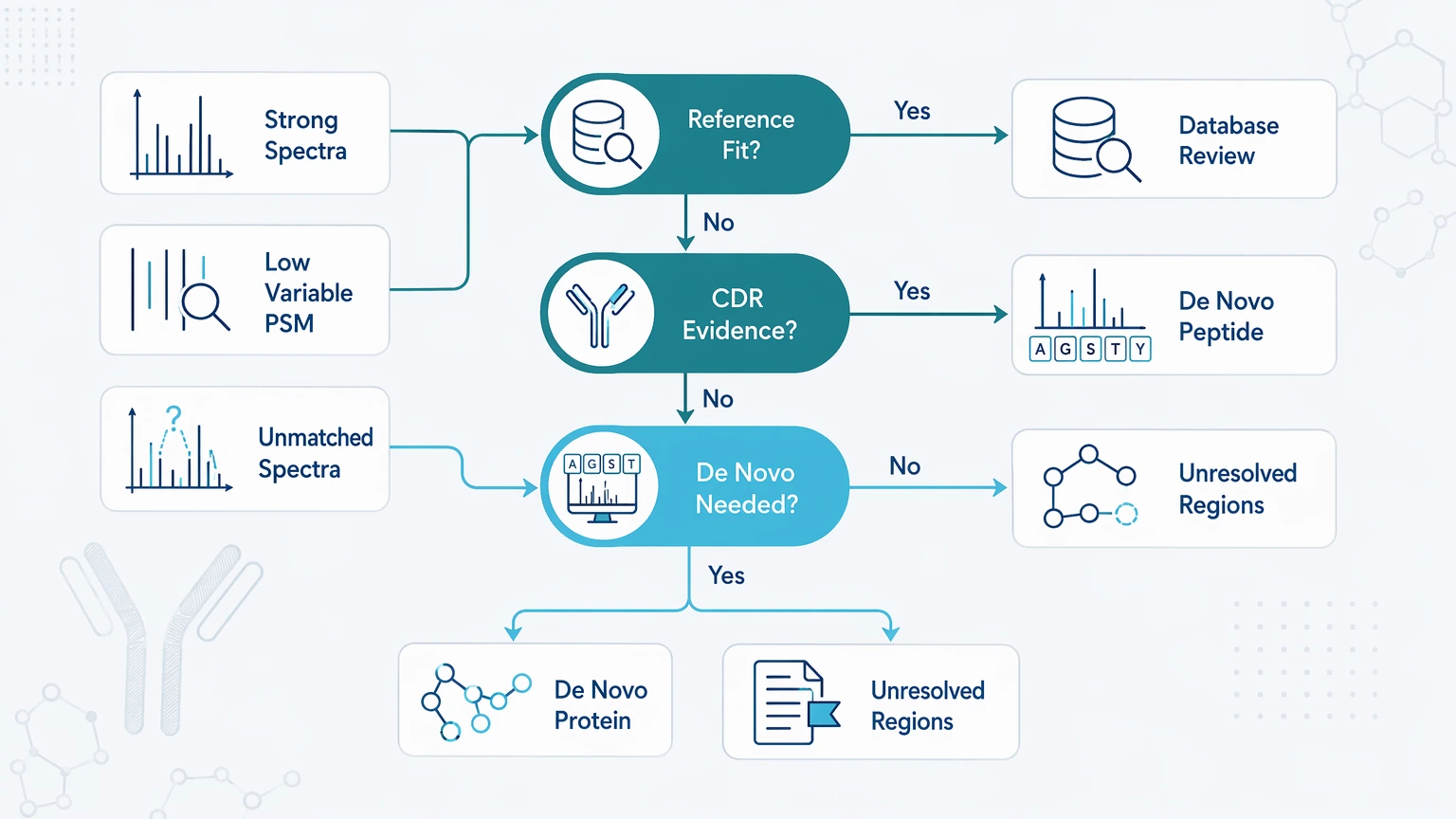
The key question is whether the database search failed in the regions that determine clonotype separation or specificity claims.
Step 5: Reset Interpretation When PTMs or Partial Evidence Are Driving the Conflict
Even a technically sound dataset can mislead if the interpretation model is too aggressive. PTM / post-translational modification burden, especially oxidation, deamidation, glycation, and glycosylation heterogeneity, changes peptide masses and fragmentation behavior. That can reduce database matches, complicate peptide assembly ambiguity, and weaken the link between sequence evidence and biological meaning.
This matters most when specificity interpretation conflicts with orthogonal assays or when CDR evidence is present but incomplete. Practical interpretation rules are straightforward: separate confirmed, ambiguous, and unresolved regions in the report; use orthogonal validation when a sequence-level difference is being linked to target binding or candidate ranking; avoid specificity claims based only on shared framework peptides; and treat candidate exclusion, candidate ranking, and provisional sequence recovery as different confidence levels.
Service Routes to Consider
When the troubleshooting question has narrowed to sequence recovery, evidence review, or confirmation planning, these routes usually map cleanly to the next project decision.
Expected Results and Validation Methods
A troubleshooting project should define what the team can learn immediately and what still needs follow-up confirmation. Realistic first-round outputs usually include a variable-region sequence coverage summary for heavy and light chains, a list of high-confidence and low-confidence peptide assignments, a matched-versus-unmatched spectra overview, candidate discriminative peptides for clonotype discrimination, an evidence map showing where sequence confidence is strong or unresolved, and an assessment of whether database search limitation is blocking interpretation.
Follow-up confirmation often requires targeted reacquisition of priority peptides, alternate digestion to recover missing CDR-containing peptides, PTM-focused review for modified peptides affecting interpretation, orthogonal validation against binding or functional data, and intact-mass or targeted LC-MS/MS confirmation for distinguishing features. If the dataset suggests that de novo-supported interpretation is justified but the evidence still needs prioritization, submit your requirements to MtoZ Biolabs with the sample type, reference availability, digestion history, and raw-data status so the team can map likely deliverables to the appropriate AAE-nanoLC-MS/MS Antibody Coverage Analysis Service, LC-MS/MS Analytical Service, or downstream validation path.
Key Cautions and Practical Limits
Very low input, poor enrichment, or severe degradation can prevent reliable variable-region recovery even when instrument data look acceptable. One run rarely settles a borderline interpretation, so replicate digestion, replicate acquisition, or an orthogonal comparison is often needed when the conclusion rests on a few peptides. Carryover, reagent contamination, and prep-to-prep bias can mimic biological differences, especially in mixed antibody samples. A low PSM rate does not automatically mean novel sequence content, and unmatched spectra do not automatically justify a full de novo claim. If the immediate question is bulk mass shift, major glycoform pattern, or confirmation of one targeted site, intact-mass analysis or targeted LC-MS/MS may be more efficient than broad de novo reconstruction.
FAQ
Why does antibody diversity analysis sometimes look less heterogeneous after enrichment?
Treat the result as a ranking change until the recovery pattern is checked. A useful follow-up is to compare pre-enrichment material, flow-through, and enriched fraction for a small set of discriminative peptides. If low-abundance candidates disappear only after one capture or cleanup condition, the result may reflect handling selectivity rather than a true change in antibody complexity.
How can I tell whether the problem is sample-related or interpretation-related?
Use one low-cost confirmation before changing the interpretation model. Intact-mass review, a replicate digest, or a targeted check of a few discriminative peptides can show whether the same candidates are physically present. If that small confirmation is stable but the database-based explanation still conflicts, the interpretation strategy is more likely the limiting factor.
Which metrics matter most when two similar antibody candidates cannot be separated?
Ask whether each candidate has independent evidence that would survive removal of shared peptides. The most useful review is a short candidate-by-candidate table with discriminative peptide sequence, spectrum quality, chain context, modification status, and whether the peptide was observed in more than one acquisition or digestion condition.
When should I repeat digestion instead of reanalyzing the same raw files?
Repeat digestion when a different enzyme could create new evidence, not simply because confidence is low. A practical trigger is a clear missing region that would be covered by an alternate cleavage pattern or a modified peptide that needs a shorter, cleaner fragment. If the same raw files already contain strong but unmatched spectra, interpretation review may come first.
Conclusion
Antibody diversity and specificity analysis troubleshooting works best when the team moves through failure points in order: sample integrity and enrichment bias, peptide generation, LC-MS/MS evidence quality, then reference fit and interpretation boundaries. That sequence helps separate true biological differences from artifacts such as contaminant background, weak CDR detectability, reference-sequence incompleteness, or overconfident reading of partial spectra. For discovery pools, engineered antibodies, PTM-rich samples, or mixed candidates where the central question is whether current data can support sequence-level diversity or specificity claims, the next step is usually a bounded review of what can be confirmed now, what needs targeted validation, and whether de novo-supported analysis is justified. If your team needs to choose between repeating the workflow and escalating the interpretation strategy, prepare the sample context, raw-data status, and decision question, then contact us for a consultation focused on practical deliverables and the most suitable next experiment.
Related Services
How to order?

