





Antibody Amino Acid Sequencing: What You Can Recover From a Purified Antibody Sample
- the VH and VL assemblies are broadly supported by peptide evidence
- key complementarity-determining region positions are observed directly
- chain assignment is consistent across digests
- remaining ambiguities are limited to positions that can be resolved through targeted follow-up
- unresolved residues within CDR3
- inconsistent peptide evidence between digests
- evidence for multiple antibody species
- uncertainty around modified or clipped regions
- a requirement to confirm a final expression construct rather than screen a candidate sequence
A purified antibody sample can often support antibody amino acid sequencing even when the original DNA, RNA, or cell source is no longer available. In many projects, the most recoverable and decision-relevant output is the variable region, including at least part of the complementarity-determining region and often broader VH and VL sequence evidence. Recovery can sometimes extend across much of the heavy chain and light chain, but exact full-chain reconstruction from protein-only material is still more difficult and should be interpreted with close attention to sequence coverage, residue ambiguity, and confidence annotations.
That distinction matters for teams working with a legacy monoclonal antibody, an externally transferred reagent, or an archived sample with incomplete records. A purified protein still carries direct amino acid evidence, but it does not preserve the original nucleotide sequence or automatically confirm native chain history. The practical question is not simply whether sequencing can be done. It is whether the recovered sequence is detailed enough for CDR review, archive recovery, recombinant re-expression, or an orthogonal validation plan.
What You Can Realistically Recover From Protein-Only Input
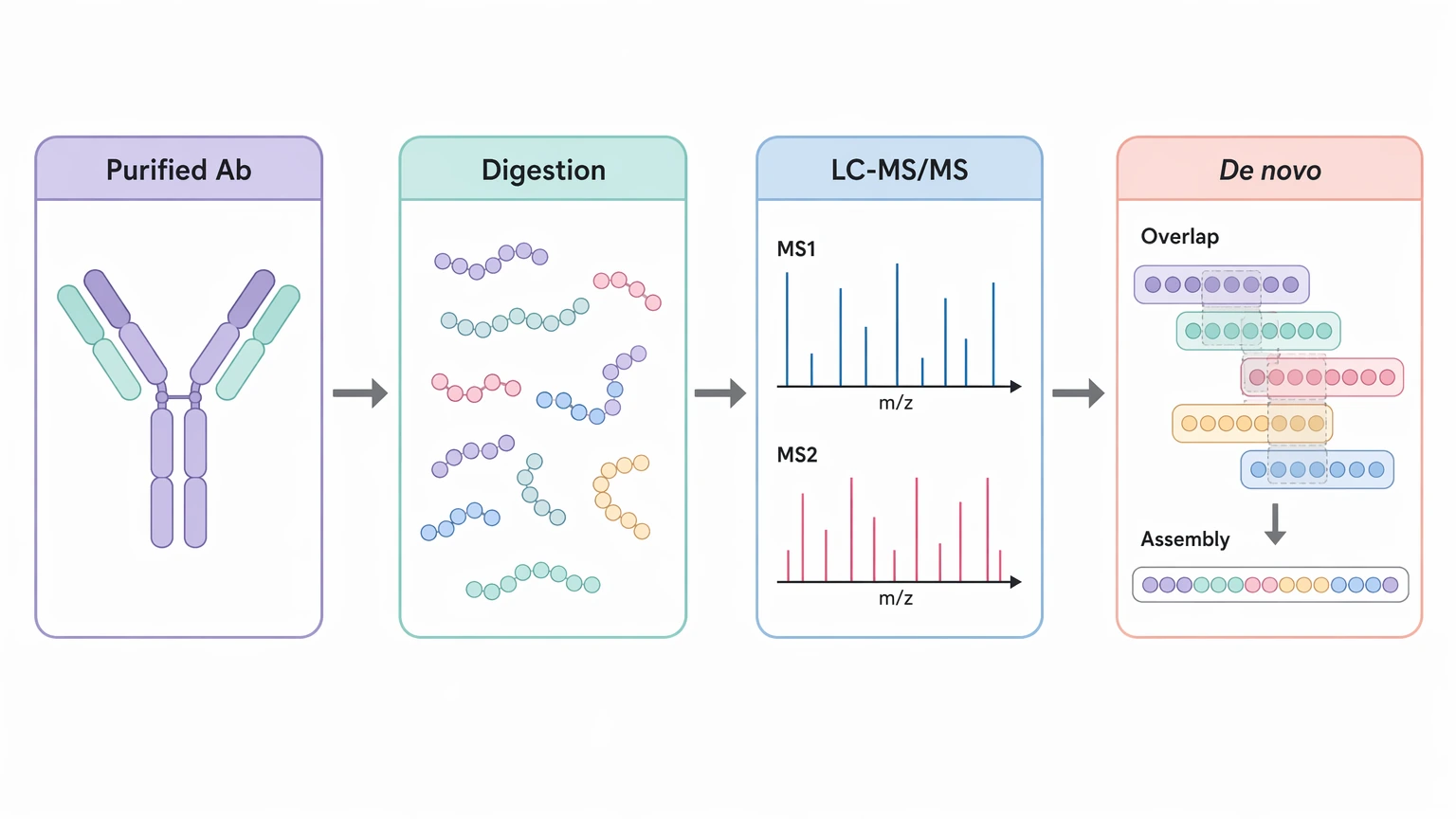
In this setting, antibody amino acid sequencing means reconstructing sequence information from the antibody molecule itself. The workflow usually combines proteolytic digestion, LC-MS/MS, de novo sequencing, and sequence assembly from overlapping peptides.
For most teams, the useful recovery levels fall into four tiers:
| Recovery target | What is often possible | Main interpretation limit |
|---|---|---|
| Complementarity-determining region evidence | Direct peptide support for parts or all of CDR1 / CDR2 / CDR3 | Loop coverage may be uneven |
| VH and VL reconstruction | Strong candidate variable region sequence with confidence flags | Some residues may remain ambiguous |
| Broader heavy chain and light chain sequence | Substantial framework and constant-region recovery | Modified or low-coverage regions can interrupt continuity |
| Exact full-chain amino acid sequence | Sometimes approachable, not automatic | Isoleucine/leucine ambiguity, missing peptides, and PTMs can block full certainty |
This hierarchy is useful because it sets expectations early. If the project goal is antigen-binding region review, some chain-level ambiguity may still be acceptable. If the goal is codon design for re-expression, unresolved positions become a much bigger issue.
Why Purified Antibody Can Still Be Informative
A purified antibody sample remains informative because the protein itself retains the amino acid order that LC-MS/MS can probe after digestion. Even without hybridoma cells or plasmids, peptide fragments can reveal conserved motifs, framework anchors, and variable-loop content that support sequence reconstruction.
Antibodies are not just another protein class, though. Their most decision-relevant information often sits in the variable region, where diversity is highest and peptide interpretation can be less straightforward. That is why antibody-focused sequence analysis needs to do more than identify peptides. It also needs to support chain assignment, distinguish VH and VL, and show how peptide evidence connects across the assembly.
How the Workflow Reconstructs Sequence
The workflow starts with sample review. Analysts first ask whether the material is likely a single purified antibody species or whether excipients, carrier proteins, degradation, or co-purified material could complicate interpretation.
Next comes chain preparation and digestion. The antibody is commonly reduced to separate the heavy chain and light chain, then digested with one or more enzymes. A multi-enzyme strategy often improves overlapping peptides, which matters because sequence assembly depends on peptide connectivity rather than isolated identifications.
The resulting peptides are then analyzed by LC-MS/MS. Fragmentation spectra support peptide interpretation, including de novo sequencing when no reliable reference is available. In antibody projects, repeat acquisitions or complementary conditions can be useful when modified peptides or low-abundance variable-region peptides are hard to capture in a single run.
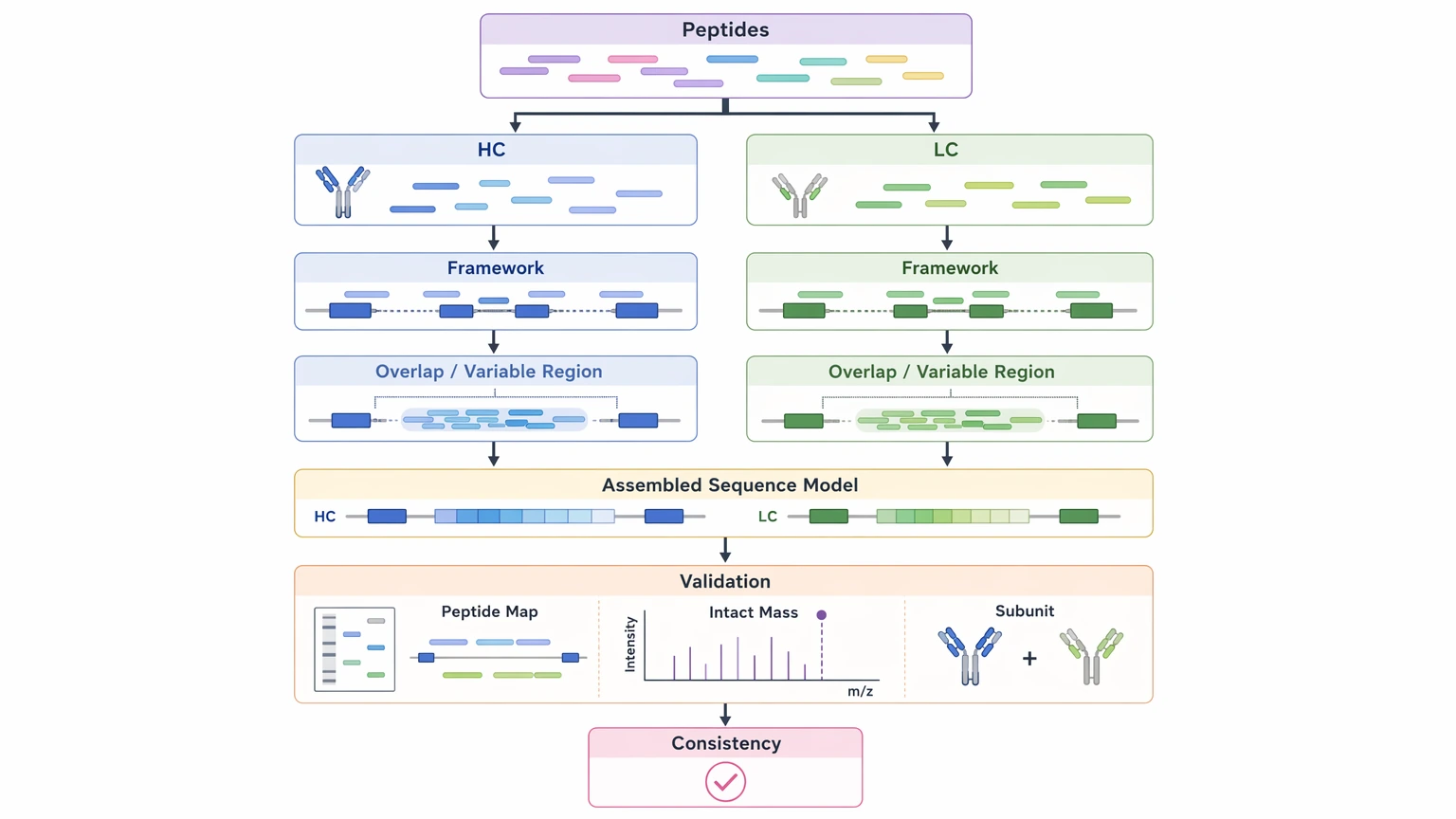
After that, the data move into assembly. Candidate peptides are grouped by chain assignment, aligned within framework anchors, and extended across the variable region wherever overlap allows. Peptide mapping, intact mass analysis, and subunit analysis can then test whether the assembled model is consistent with the observed molecule.
The Four Constraints That Most Often Limit Recovery
Not every challenge in protein sequencing matters equally for this decision. For a purified-antibody sequencing project, four categories usually shape the outcome.
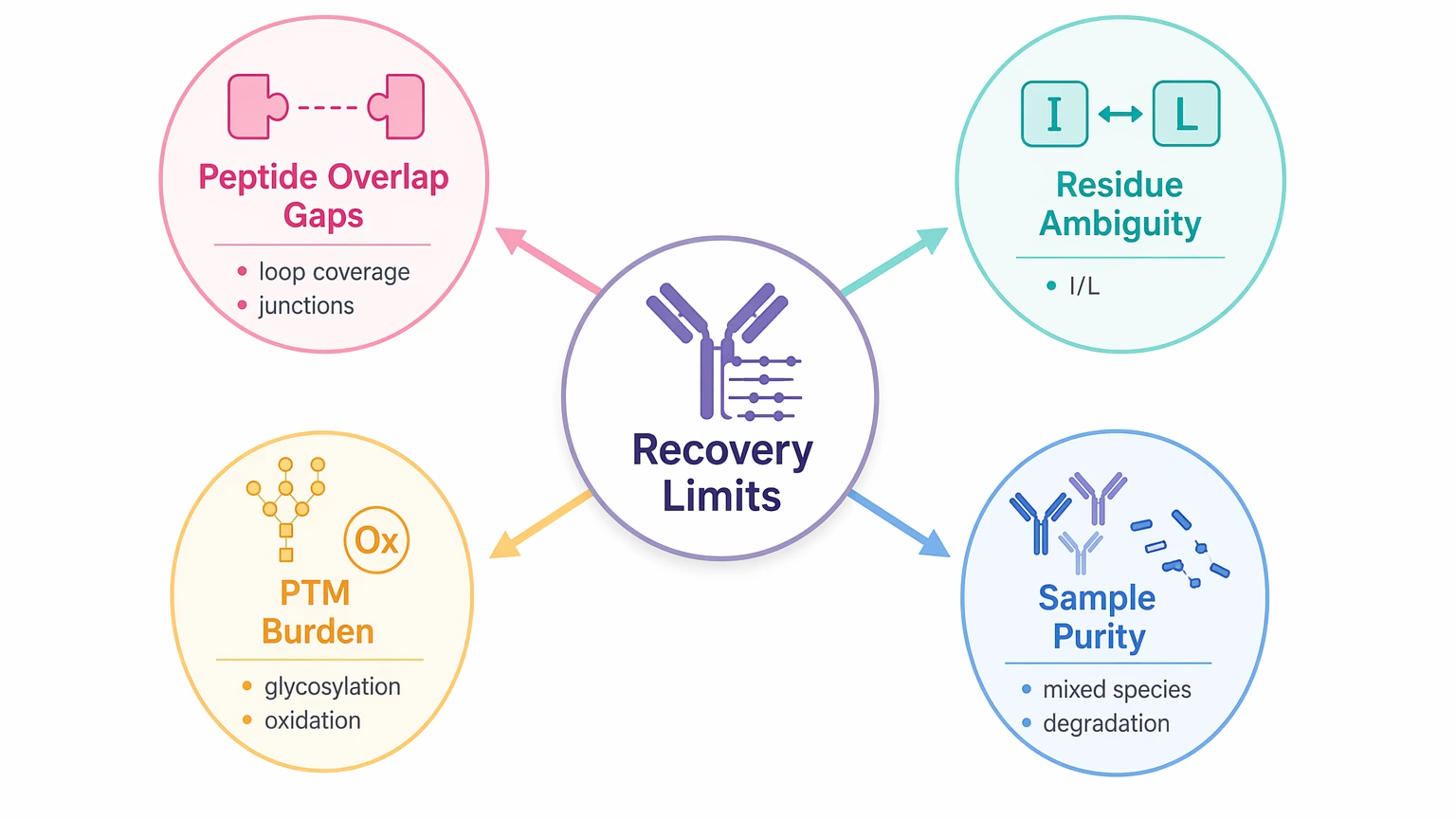
1. Peptide overlap gaps
A clean peptide spectrum is not the same as a complete sequence. If peptides do not overlap well, the report may contain confident local sequence blocks but weaker long-range sequence assembly. This tends to matter most in hypervariable loops and at peptide junctions.
2. Residue-level ambiguity
The best-known example is isoleucine/leucine ambiguity. Because those residues are isobaric, MS-based evidence does not always distinguish them directly. Some ambiguities can be narrowed through overlap logic or complementary evidence, but not every position can be fully resolved from protein data alone.
3. Post-translational modification burden
A post-translational modification can complicate fragmentation and peptide interpretation. In antibodies, glycosylation in the Fc region is a common source of complexity. Oxidation, deamidation, pyroglutamate formation, and clipping can also shift mass patterns and make affected peptides harder to read cleanly.
4. Sample composition and purity
A “purified” label does not always mean a single intact analyte. Formulation components, partial degradation, mixed antibody species, or contamination can reduce sequence confidence and make chain assignment more difficult. When the project starts with a limited or heterogeneous sample, expectations should be anchored to what the peptide evidence actually supports.
How to Read a Sequencing Deliverable
A good sequencing report should not present a single sequence string without context. It should show which parts of the reconstruction are directly observed, which are inferred, and where confidence starts to drop.
Look for these reporting elements:
| Deliverable feature | Why it matters for downstream decisions |
|---|---|
| Sequence coverage | Shows which regions have direct peptide support |
| Chain assignment | Confirms whether peptides map to heavy chain or light chain |
| CDR recovery confidence | Indicates whether antigen-binding loops are supported by direct evidence |
| Residue ambiguity flags | Identifies positions that may block final construct design |
| Agreement with intact mass analysis or subunit analysis | Tests whether the assembled sequence matches the observed antibody species |
This is also the stage where project intent should guide interpretation. For archive recovery, a report with a few framework ambiguities may still be useful. For construct synthesis, the more important question is whether unresolved residues fall in CDRs, termini, or other function-sensitive positions. If your team is deciding whether a protein-only dataset is strong enough to move into rebuilding, you can submit your requirements and evaluate your project with MtoZ Biolabs using the sample format, intended use, and any prior analytical data as the decision context.
When the Output Is Often Sufficient for Re-Expression Planning
Recovered amino acid sequence can support early recombinant re-expression planning when several conditions line up:
That still does not mean the project is ready for an immediate final construct lock. Re-expression planning is strongest when the sequence package is paired with a validation strategy for sensitive residues, expected modifications, and identity confirmation after expression.
When to Add Orthogonal Validation or a Nucleic-Acid-Based Route
Protein-only sequencing answers many practical questions, but not all of them. Add orthogonal validation when the downstream decision is sensitive to local ambiguity or when the assembled model needs stronger support.
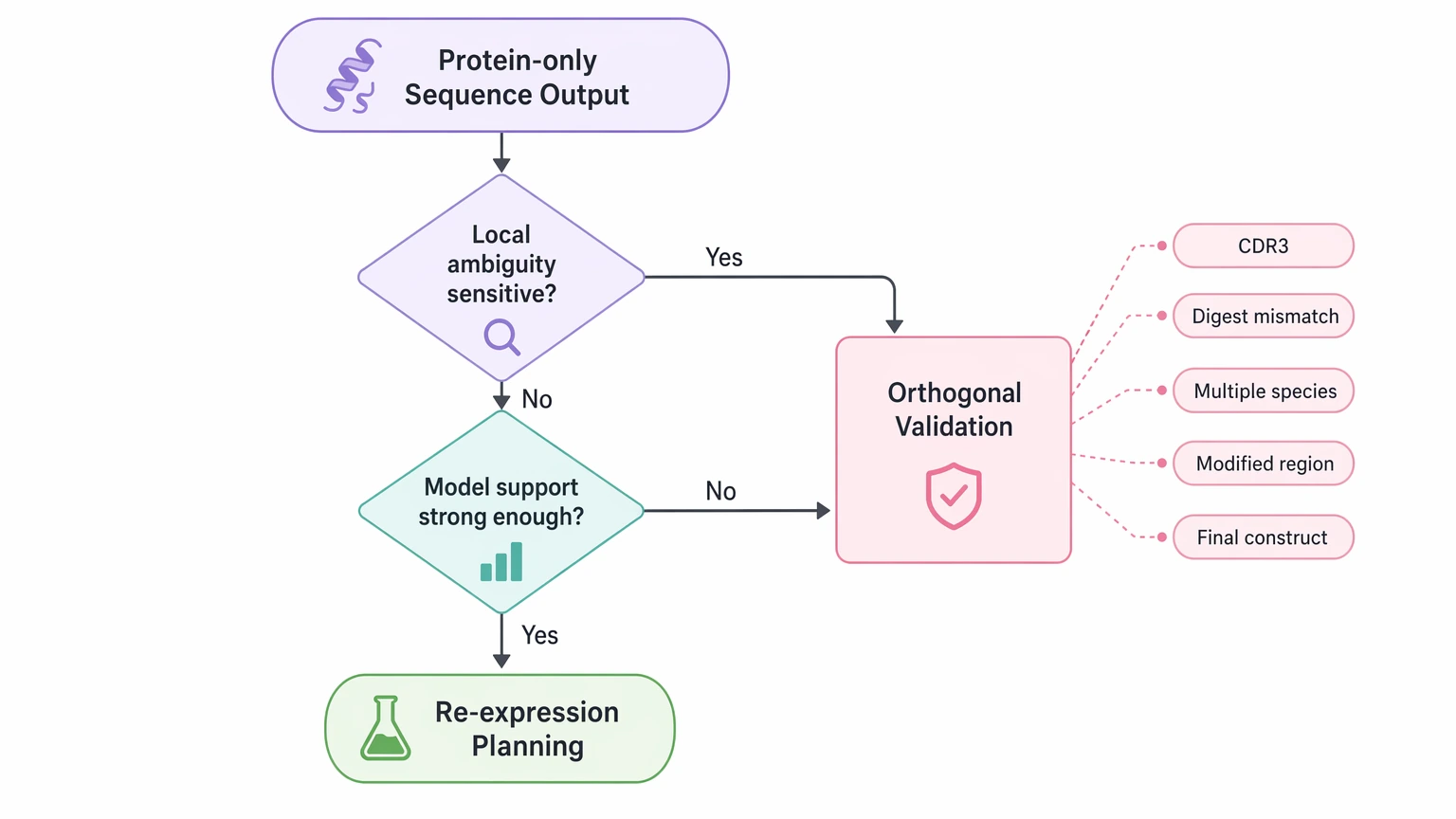
Common triggers include:
If viable cells, hybridoma material, or nucleic acid templates become available, RACE or NGS can resolve questions that protein data alone may leave open. In that setting, the protein-level result still matters because it provides an independent sequence view and helps test whether the recovered coding sequence matches the actual antibody material.
Typical Project Scenarios
A legacy discovery program may retain only a vial of purified IgG with no trustworthy plasmid archive. In that case, antibody amino acid sequencing can clarify whether the sample still supports variable-region recovery and whether the result is strong enough to justify rebuild work.
A second common case is partner material transfer. A team receives a purified antibody with partial documentation and needs to know whether it behaves like a single analyte and whether the variable region sequence is defined well enough for internal comparison or archiving.
A third case is pre-rebuild triage. The team already expects re-expression to be the likely next step, but it needs to decide whether the current protein sample can support that effort or whether the project should shift toward nucleic-acid recovery first.
Final Takeaway
A purified antibody sample can often yield decision-useful antibody amino acid sequencing output, especially at the complementarity-determining region, VH and VL, and broader heavy chain and light chain levels. What matters is not a blanket claim of “full sequencing,” but whether the observed peptide evidence supports the level of confidence your project actually needs. For legacy antibody recovery, partner sample review, or sequence-guided rebuild planning, the most practical next step is to define the intended use, map unresolved risks to the report structure, and then contact MtoZ Biolabs to submit your requirements and discuss the sequencing and validation workflow before committing to a construct decision.
FAQ
Can antibody amino acid sequencing confirm the original clone source?
No. It can recover protein-level amino acid information from the antibody you have, but it does not prove the original clone lineage, vector design, or biosynthetic source.
Is a reduced heavy-chain and light-chain workflow always required?
Not always, but reduction often improves chain assignment and simplifies interpretation. For some samples, intact and subunit views are also useful because they show whether the assembled peptides match the antibody mass pattern.
If framework regions look complete, can CDR3 still remain uncertain?
Yes. Framework peptides may assemble well while CDR3 remains weaker because of peptide length, fragmentation behavior, or limited overlap in that loop.
Does formulation buffer always need to be removed before LC-MS/MS?
Not always completely, but excipients and stabilizers can interfere with digestion or ionization. The need for cleanup depends on what is present in the formulation and how much sample is available for repeat work.
Can a polyclonal preparation be handled the same way as a purified monoclonal antibody?
No. Mixed antibody populations create major interpretation problems because peptide evidence comes from multiple related molecules. This article’s recovery logic is most applicable to a purified monoclonal antibody.
What is the most useful first question to ask before starting?
Ask what downstream decision the sequence must support. Archive documentation, CDR inspection, and final construct design do not require the same level of sequence confidence.
How to order?

