





Why De Novo Peptide Sequencing Software Misses Low-Intensity Spectra and What to Check Before Reprocessing
Reprocessing is worth trying only when the missing low-intensity spectrum still carries fragment evidence that the original workflow weakened, misassigned, or filtered away. If the tandem mass spectrum is sparse, mixed, or lacks enough b-ion series or y-ion series continuity to support a believable path, de novo peptide sequencing software is usually running into an information limit, not missing an obvious answer.
Quick Decision Check
Use this short triage block before starting another run:
| What you observe | Most likely interpretation | Better next step |
|---|---|---|
| The scan is present in the raw file but weakened or altered in mzML or MGF | File conversion, centroiding, peak picking, or denoising changed what the engine sees | Re-export and reprocess |
| The precursor ion is visible, but charge-state assignment or monoisotopic precursor annotation looks unstable | Precursor handling is distorting mass interpretation | Correct precursor settings before rerunning |
| The raw MS/MS spectrum remains sparse, noisy, or chimeric across formats | The scan may not contain enough fragmentation coverage for de novo peptide sequencing | Consider reacquisition or orthogonal validation |
| Lowering score thresholds adds more calls but not better sequence confidence | The issue is ambiguity, not simple filtering | Review sequence tag quality rather than output count |
This article centers on one practical decision: should your team expect better sequence recovery from reprocessing alone, or does the missing output reflect a de novo-specific interpretation limit?
Where This Problem Usually Appears
This problem usually shows up after a first-pass de novo peptide sequencing run on LC-MS/MS data from unknown peptides, PTM-rich samples, degradation products, natural extracts, or low-input digests. An analyst checks the raw file, sees a precursor ion and a visible MS/MS spectrum near the expected retention time, then finds that the report contains few sequence tags, weak sequence confidence, or no usable call for that scan.
That mismatch matters more in de novo peptide sequencing than in a database-assisted workflow. A weak tandem mass spectrum may still contribute to a database search when the algorithm can anchor interpretation to a known candidate. De novo peptide sequencing software does not have that fallback. It has to reconstruct amino acid order from fragment evidence alone, so missing ladder continuity hurts much more.
Why Low-Intensity Spectra Fail in De Novo Interpretation
A low-intensity spectrum is not automatically useless, but it often fails in exactly the way de novo interpretation cares about most. The issue is not only that the peaks are small. The bigger problem is that too few fragment ion peaks survive above the noise threshold to support a consistent mass-difference path across the peptide.
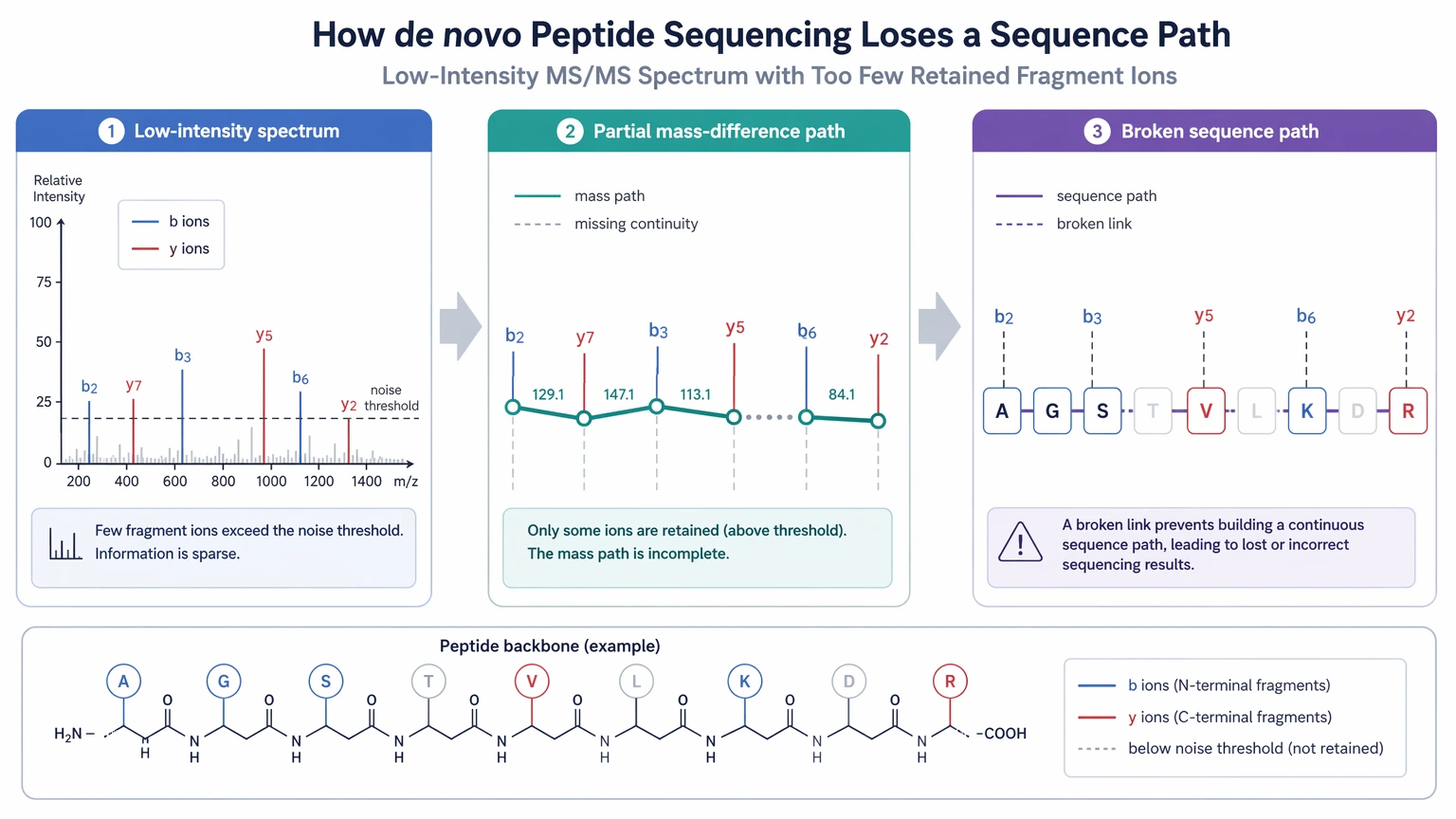
That can happen in a few ways. The spectrum may contain too few fragment ions overall. It may have a fair number of peaks, but not enough continuity in the b-ion series or y-ion series. It may also include fragments from more than one precursor ion because of co-isolation interference, creating a chimeric spectrum that looks busy but remains hard to interpret. PTMs make this even harder because unexpected mass shifts can break a path that was already weak.
So a visible scan is not the same thing as a sequence-informative scan. That distinction should shape the rest of your troubleshooting.
The Four Checks That Matter Most Before Reprocessing
For this problem, the highest-value root causes usually fall into four groups. Going much wider than that often burns time because the real question is simple: does the acquired scan still hold sequence evidence that can be recovered?
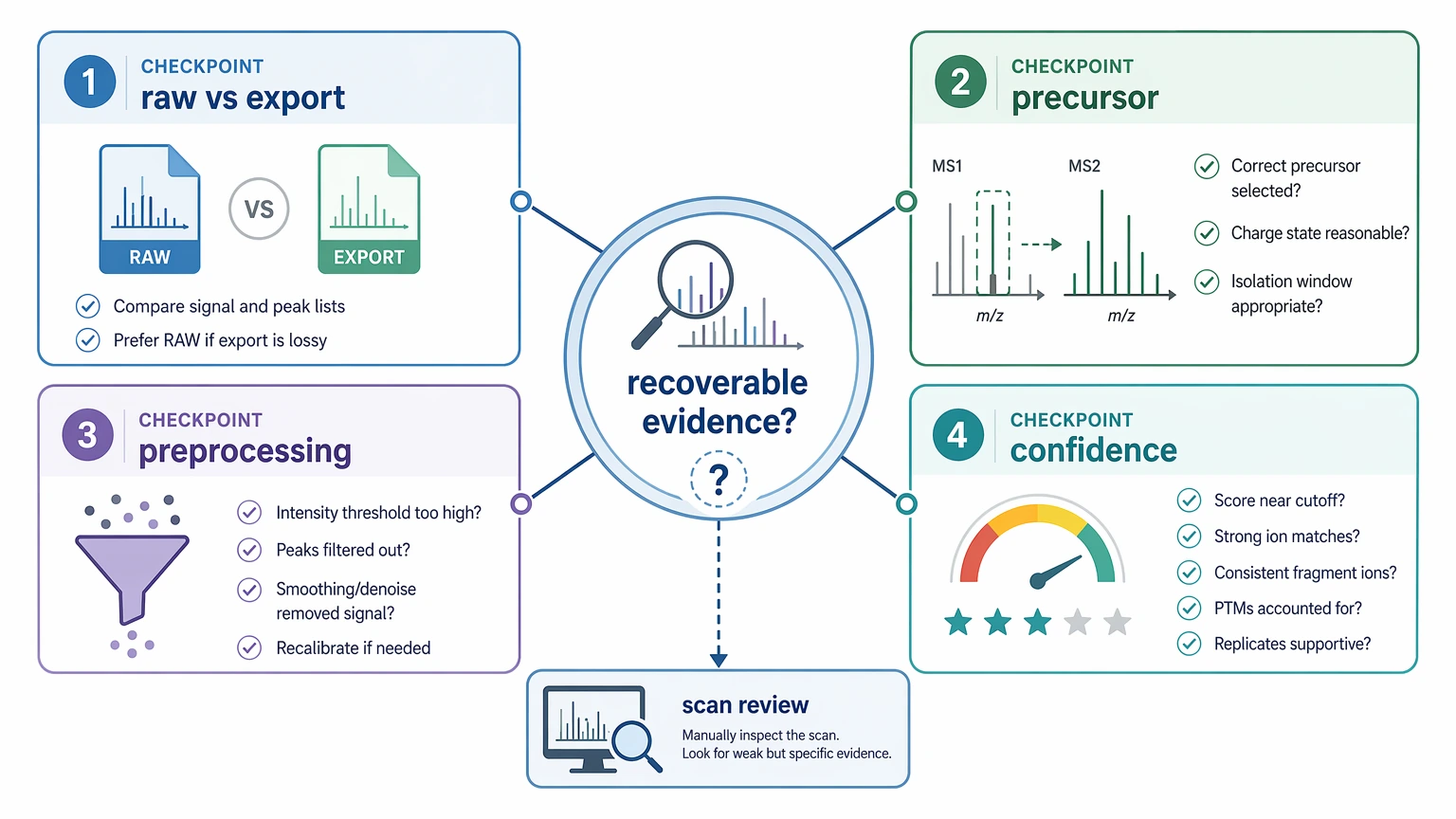
1. Confirm raw-file presence and export integrity
Start by comparing the vendor raw file with the mzML or MGF that went into the de novo engine. Confirm that the same scan number, precursor metadata, and major fragment ion pattern appear in both. If the exported file shows fewer peaks, shifted precursor annotations, or a changed intensity profile, the software may be processing a different version of the spectrum than the one you reviewed by hand.
Before the table below, ask one direct question: did the spectrum survive file handling in a form that still supports de novo interpretation?
| Scenario | Recommended workflow | Key limitation | Validation need |
|---|---|---|---|
| Raw file shows the scan, exported file does not | Re-export mzML or MGF with revised settings | Lost peaks are not recoverable from the old export | Compare scan presence and precursor metadata |
| Raw and export both show the scan, but peak density changes sharply | Recheck centroiding and peak picking | Weak fragment ions may have been compressed or removed | Overlay peak count and major fragment positions |
| Raw, mzML, and MGF agree closely | Move to precursor and scoring review | Export changes are unlikely to explain the loss | Inspect charge-state assignment and score filtering |
The point here is plain enough: if export stripped away weak but real fragment ion peaks, another de novo run on that same damaged peak list will not fix the problem.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
2. Verify precursor interpretation
Low-intensity spectra often sit near the edge of stable precursor characterization. That is why monoisotopic precursor assignment and charge-state assignment matter so much. A small precursor error can send the engine down the wrong mass path, and sparse fragment evidence may not be strong enough to pull it back.
Also check isolation quality. A broad isolation window or nearby precursor crowding can introduce co-isolation interference. In that case, the precursor ion may be real, but the tandem mass spectrum reflects mixed fragment populations. Reprocessing may still help when precursor correction is the main issue, but it will not fully rescue a truly chimeric spectrum.
3. Check whether preprocessing removed the only useful evidence
This step often decides whether reprocessing is worth the effort. Compare the number of retained peaks after centroiding, deisotoping, and denoising. On weak scans, those steps can reduce clutter, but they can also remove the few peaks that support a real sequence tag.
Use the table below to judge whether preprocessing changed the data enough to justify another run.
| Evidence | What it supports | Limitation | Follow-up |
|---|---|---|---|
| Weak peaks disappear after centroiding or denoising | Reprocessing may recover useful sequence evidence | Noise may increase as well | Regenerate the peak list with milder filtering |
| Precursor metadata stays stable, but MGF peak count drops sharply | Export settings are suppressing informative ions | Some engines still need simplified input | Test a second export path |
| Fragmentation coverage stays poor in raw and processed forms | The scan is nearing a true information limit | Software cannot create fragment ions that were never acquired | Shift toward reacquisition or validation |
The practical takeaway is simple: reprocessing helps when the pipeline hid evidence that was already there. It does not help when that evidence was never acquired.
4. Review confidence logic before lowering every threshold
When sequence yield looks disappointing, many teams lower confidence score cutoffs first. That can increase the number of reported calls, but not necessarily their usefulness. Review how your de novo peptide sequencing software defines confidence score, local confidence, or sequence confidence. A scan may be excluded because several near-equal paths fit the same sparse evidence.
This is also where PTMs and database search limitation become relevant. A modified peptide with weak fragmentation can generate several plausible paths that differ because of missing ions or shifted masses. Any sequence call recovered under those conditions should be treated as provisional. In PTM-rich or unknown-sequence work, one limitation is hard to avoid: low-intensity MS/MS interpretation may yield only partial sequence tags, and follow-up confirmation is often needed before treating a candidate as a supported peptide assignment.
Step-by-Step Reprocessing Strategy
Once the four checks above are complete, use a troubleshooting sequence instead of a generic rerun.
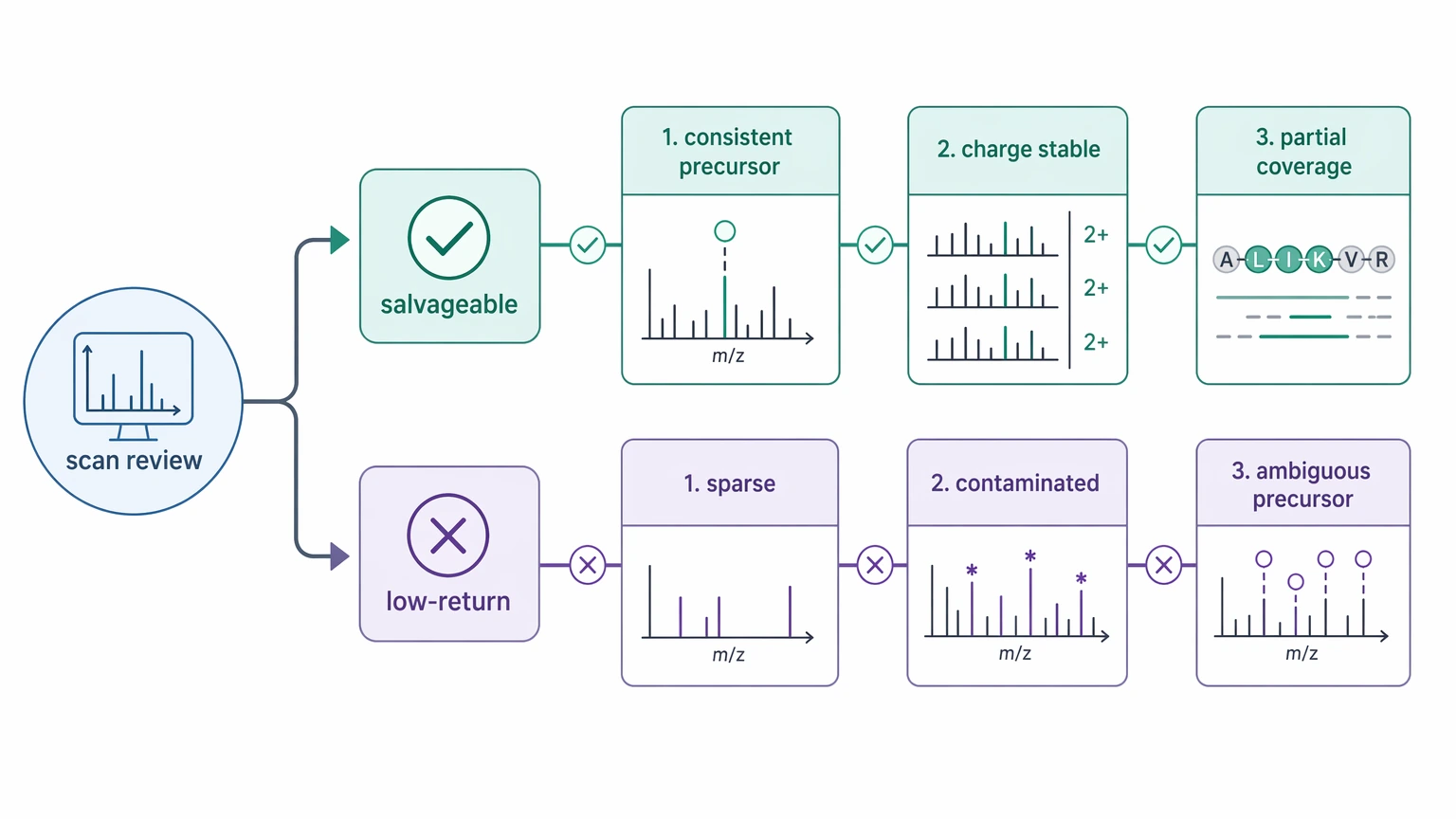
First, separate scans into salvageable and low-return groups. Salvageable scans usually show consistent precursor ion detection across files, plausible charge-state assignment, and at least partial fragmentation coverage on manual review. Low-return scans usually remain sparse, contaminated, or precursor-ambiguous no matter which file view you use.
Second, define exactly what will change in the rerun. Useful changes include a cleaner export path, less aggressive denoising, corrected monoisotopic precursor handling, revised mass tolerance, or PTM-aware interpretation when the sample context supports it. Random parameter changes rarely answer the real decision question.
Third, compare rerun output at the scan level, not only by total sequence count. If the same precursor ion gains a longer sequence tag, better local confidence, or clearer ladder continuity, the rerun is doing something useful. If output count rises but sequence confidence does not, the rerun may only be surfacing weaker guesses.
If your team still has uncertainty after raw-file review, export review, and score interpretation, you can submit your requirements to MtoZ Biolabs to evaluate the project data path and decide whether de novo peptide sequencing reprocessing, LC-MS/MS review, or a different sequencing strategy is the better next move.
Expected Results and Validation Methods
A justified reprocessing attempt should produce immediate deliverables that are visible before any broader biological interpretation. Those outputs can include a regenerated mzML or MGF, revised scan retention counts, corrected precursor annotations, longer sequence tags on previously missed scans, or an improved confidence score distribution for the same precursor ion set.
Useful, yes. But those outputs are not the same as confirmation. Follow-up confirmation answers a different question: does the new call still look credible outside the reprocessing environment? Depending on the project, that may involve replicate consistency, targeted reacquisition, orthogonal validation, or comparison against expected PTM chemistry and fragmentation behavior.
Put another way, immediate deliverables show whether reprocessing recovered interpretive signal. Follow-up confirmation shows whether that signal is strong enough to support downstream decisions.
When Reacquisition or Another Workflow Is the Better Next Step
Reacquisition becomes the more realistic choice when the raw MS/MS spectrum stays sparse across formats, precursor assignment remains unstable, or contamination risk remains high. The same is true when the analyte class does not fragment well under the original acquisition conditions or when PTM complexity overwhelms the fragment evidence that was collected.
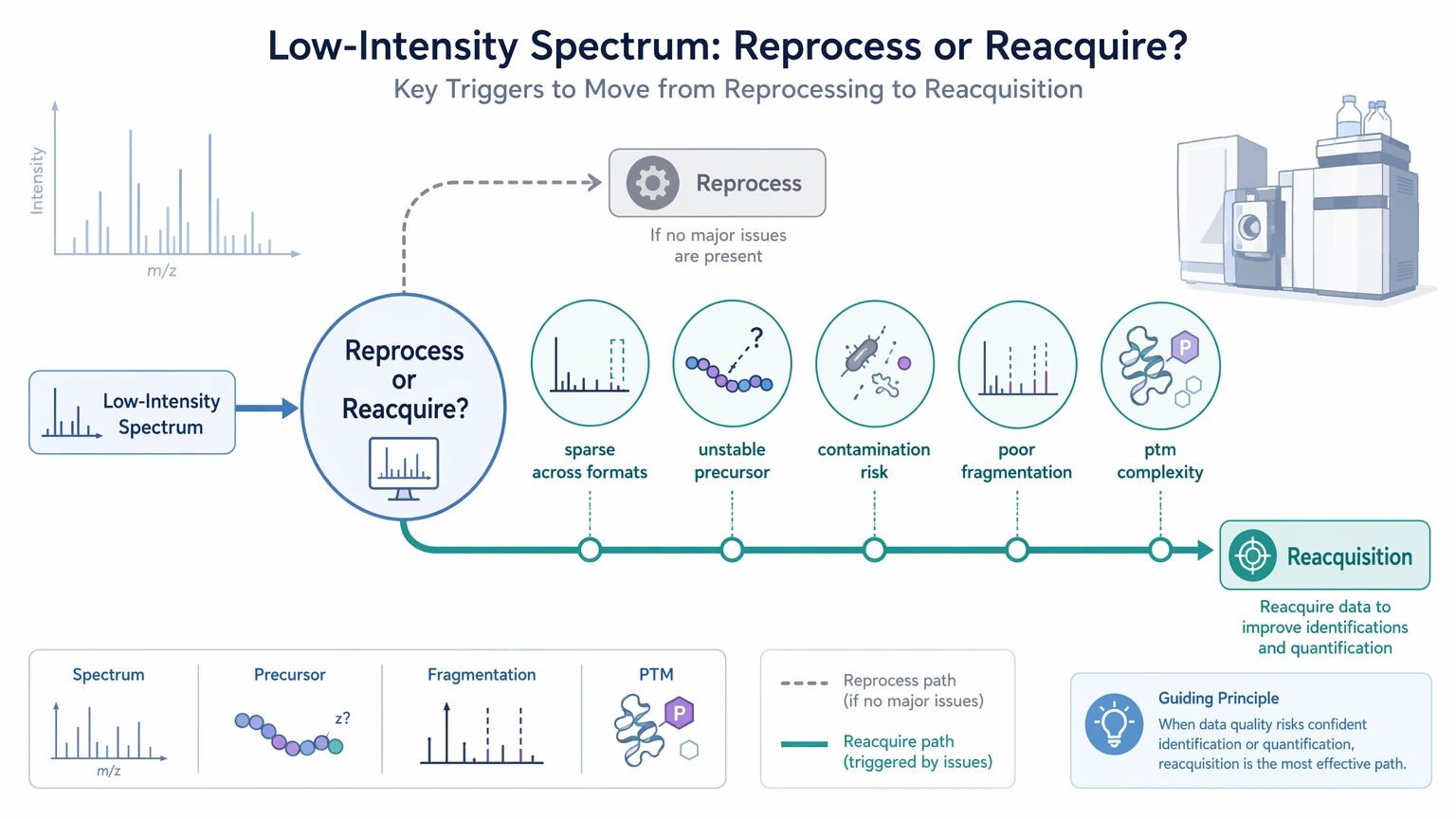
A different workflow may also make more sense when short sequence tags are enough for the next phase. In that situation, orthogonal validation, targeted MS, or escalation toward de novo protein sequencing may be more efficient than repeating full de novo peptide sequencing on unchanged low-information scans.
For labs weighing those paths, MtoZ Biolabs can review project context, sample constraints, and sequence-confidence boundaries so the next step is based on data quality and intended use rather than another software rerun. If you need that review, contact us with the sample background, acquisition details, and the scans that are failing.
Key Cautions and Practical Limits
Keep five limits in mind before treating reprocessing as the answer.
Sample quality and amount still matter. If the material is limited or degraded, you may not get many opportunities to test alternative acquisition conditions.
Controls and repeat observations matter too. A weak precursor ion that appears in replicates deserves more attention than a one-off event that never comes back.
Batch effects and contamination risk can distort low-intensity interpretation. Carryover, background ions, and mixed isolation windows can all create fragment patterns that look more interpretable than they really are.
Interpretation boundaries should stay explicit. A low-confidence de novo candidate or short sequence tag can guide follow-up work, but it does not automatically establish peptide identity.
Finally, another method or outside support may be the better next step when the main obstacle is missing fragmentation information rather than software handling. That is usually the point where targeted reacquisition, orthogonal validation, or an external review of raw files and preprocessing choices saves more time than another blind rerun.
The most useful conclusion here is usually technical, not absolute: if the scan still contains recoverable fragment evidence, careful reprocessing may improve sequence output; if it does not, the project is better served by reacquisition or a validation-focused workflow. That judgment fits unknown peptides, PTM-rich samples, and low-input LC-MS/MS studies where de novo interpretation carries real uncertainty. If you want to evaluate that boundary before spending more cycles on the same file, submit your requirements to MtoZ Biolabs and discuss the study, spectra, and reprocessing options with the team.
FAQ
Can profile-mode data help when centroided data look too sparse?
Sometimes. If the software or conversion pipeline allows a better centroiding pass from profile data, weak fragment ion structure may survive more effectively. The gain depends on whether the missing information was lost during preprocessing rather than absent at acquisition.
Why do short sequence tags sometimes matter more than full-length calls?
A short sequence tag can still narrow candidate space, guide targeted reacquisition, or support orthogonal validation planning. In low-intensity projects, partial but defensible information is often more useful than a forced full-length call with weak sequence confidence.
Does tighter mass tolerance always improve de novo interpretation?
No. Tighter mass tolerance can reduce false paths when mass accuracy is stable, but it can also remove valid peaks if calibration or precursor annotation is imperfect. The setting needs to match instrument behavior and the quality of precursor handling.
How do I tell whether a spectrum is chimeric enough to stop rerunning it?
Look for inconsistent fragment relationships, evidence of multiple precursor ion populations in the isolation window, and unstable sequence paths that change sharply with minor parameter shifts. Those patterns usually point to mixed evidence rather than a simple scoring problem.
Should I try a database search on the same scans even if the goal is de novo peptide sequencing?
It can be a useful comparison step when you suspect a known contaminant, a common digest product, or a modified form of a familiar peptide. It will not replace de novo peptide sequencing, but it can help separate true novelty from a database-search limitation or an interpretation artifact.
How to order?

