





Whole Genome Sequencing De Novo Assembly: A Planning Guide for New Organism and Non-Reference Genome Projects
- routine database search fails because the sample sequence is missing, mismatched, or heavily modified
- the fragmentation spectrum contains interpretable b ions and y ions
- the sample is clean enough to support useful peptide-spectrum interpretation
- the team accepts that the first output may be ranked candidate sequence calls or a sequence tag, not a single final answer
- spectra are weak, sparse, or inconsistent across repeats
- mixture complexity is high enough to blur fragment assignment
- the team has not defined what counts as decision-ready evidence
- short sequence tags for follow-up searching
- ranked candidates with confidence annotation
- inferred protein regions
- validation-priority targets for synthesis or targeted confirmation
- continuous stretches of b ions or y ions
- enough signal to support residue ordering rather than isolated mass differences
- consistent precursor interpretation across repeats
- mass accuracy and signal-to-noise that support confident assignment
- ranked candidate sequence calls
- annotated sequence tags
- explicit unresolved positions
- modification hypotheses
- residue-level or segment-level confidence annotation
- recommended follow-up confirmation steps
- targeted LC-MS/MS against key sequence segments
- synthetic peptide confirmation
- Edman-compatible fragment checks in suitable cases
- proteogenomic cross-checking when genome or transcript information exists
- Will the deliverable distinguish a sequence tag from a full candidate?
- How will unresolved residues be labeled?
- Will the report show evidence-supported regions versus inferred regions?
- How will PTM hypotheses and localization uncertainty be stated?
- What confirmation steps are recommended before downstream use?
- De Novo Peptide Sequencing Services
- De Novo Protein Sequencing Service
- LC-MS/MS Analytical Service
- LC-MS/MS-Based Targeted Site Validation Service
Proceed with a de novo LC-MS/MS workflow when failed identification points to a real reference database limitation, not just weak acquisition. Before kickoff, lock down four items: sample state, spectral quality, the target evidence level, and an orthogonal validation plan. If the current data support only a short sequence tag, the project may still justify moving forward, but the scope should stay at candidate discovery rather than sequence confirmation.
For unknown peptide identification and novel protein identification, the first planning question is usually not which software to try next. It is what kind of answer the project actually needs. A team preparing for synthesis, publication-critical claims, IP review, or functional testing needs stronger fragmentation spectrum continuity, better sequence coverage, and firmer confirmation than a team that only needs to narrow candidates from a non-reference sample.
Quick Decision Block
Choose de novo peptide sequencing or de novo protein sequencing when:
Pause and improve the workflow first when:
One limitation should be stated clearly at the start: standard LC-MS/MS evidence may leave unresolved positions, especially under leucine/isoleucine ambiguity, heavy post-translational modification (PTM) burden, or incomplete fragmentation, so sequence confidence has to follow the fragment support that is actually present.
When De Novo Sequencing Is the Right Escalation Beyond Database Search
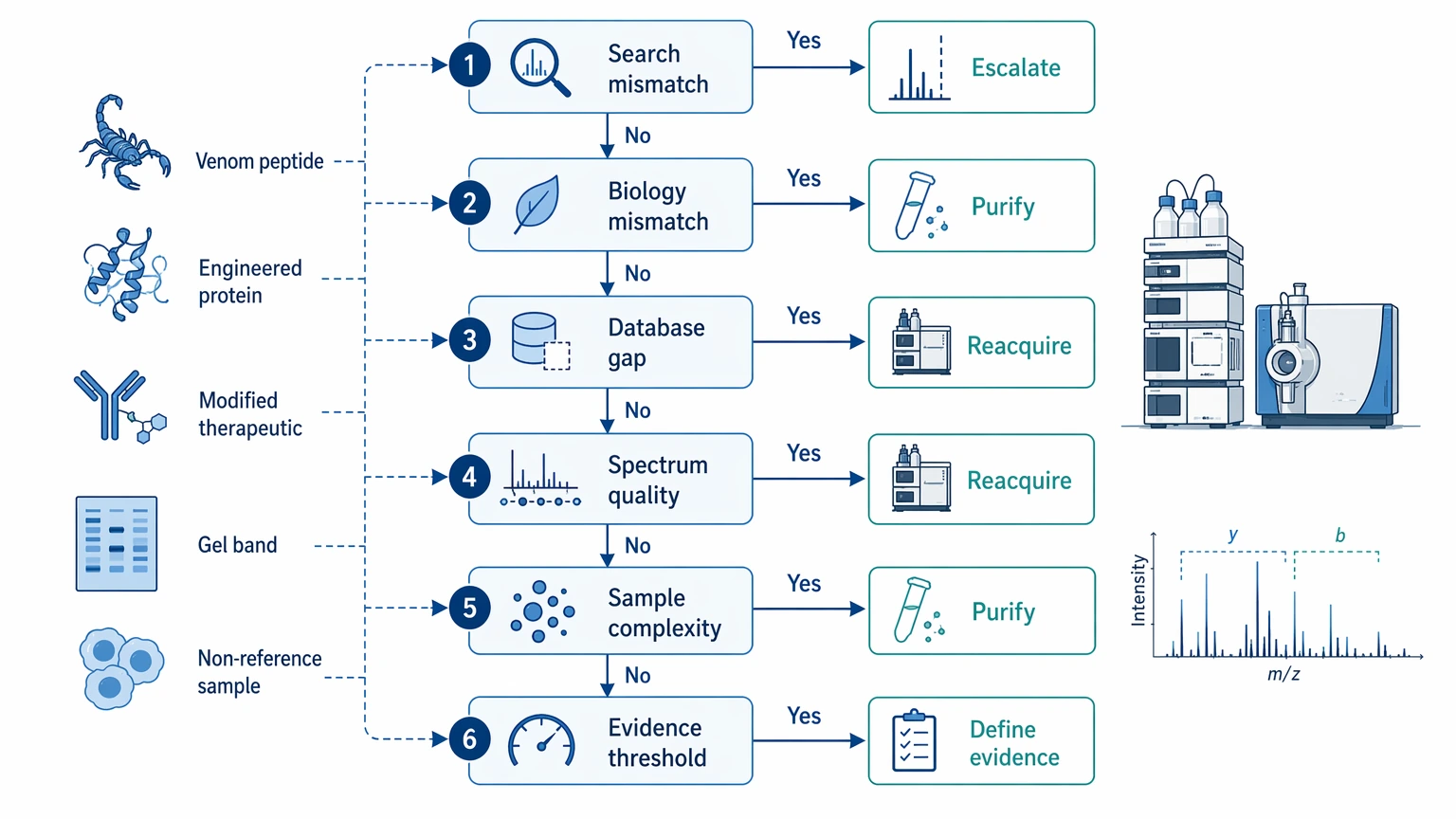
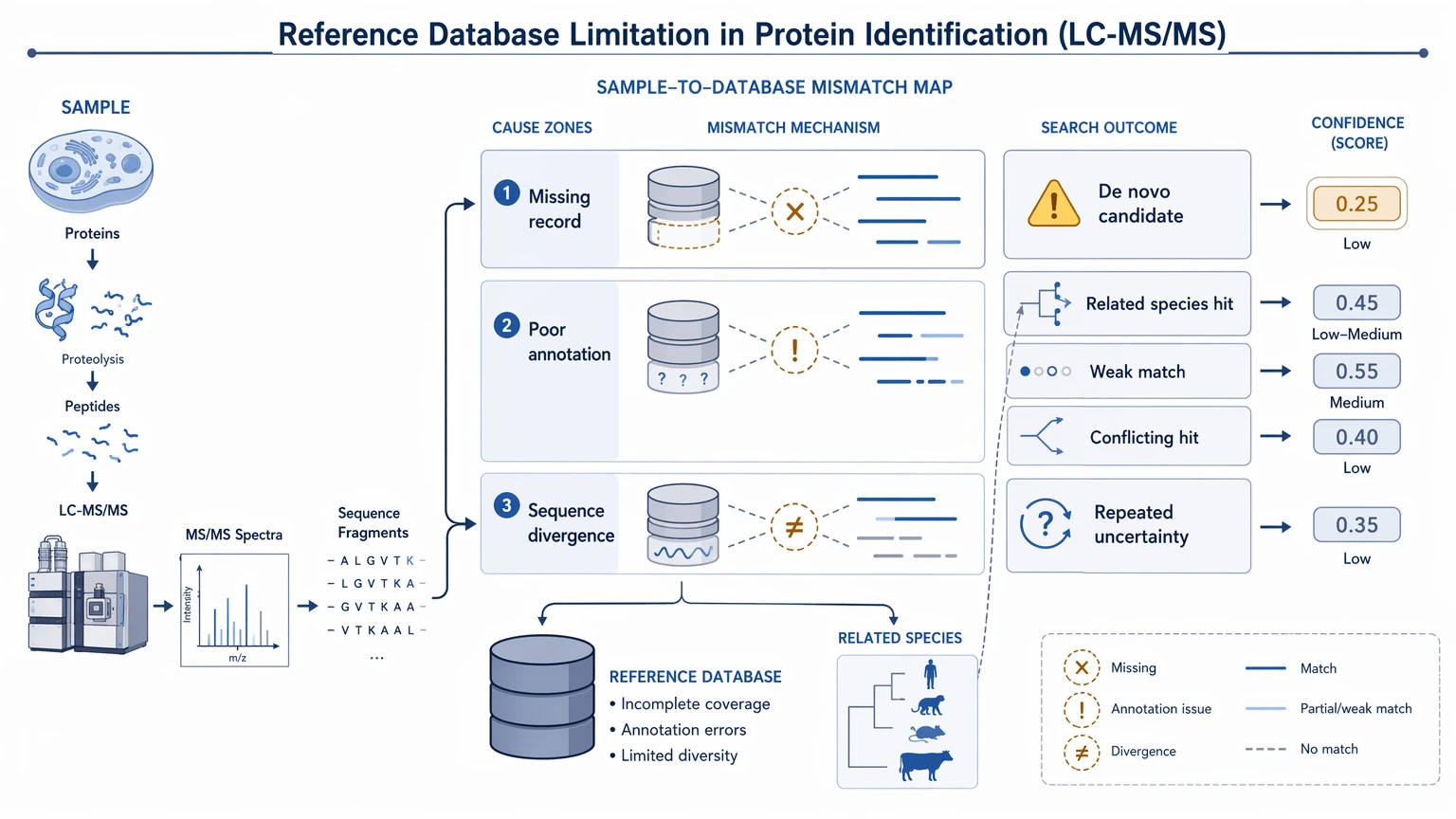
Teams usually get to this point after a database search returns weak matches, conflicting identifications, or hits to related species that do not match the biology. Common triggers include venom peptides, engineered proteins, modified therapeutics, gel bands from unexpected targets, and samples from organisms with incomplete proteome records.
That still does not mean de novo work is automatically the next move. In practice, four cause categories matter most.
1. True reference database limitation
A project is a strong candidate for de novo peptide sequencing when the biological source is missing from available records, poorly annotated, or likely to contain real sequence divergence. In that situation, another standard search often produces the same uncertainty with a different score.
2. Weak spectral quality
Some apparent de novo cases are really acquisition problems. If tandem mass spectrometry data do not show readable ion ladders, direct sequence inference will also stay weak. Better acquisition can matter more than a more aggressive interpretation algorithm.
3. Excessive sample complexity
Mixed fractions, low-abundance isolates, and partially purified digest backgrounds often break ion-series continuity. The result may be a short sequence tag with limited confidence annotation rather than a useful full candidate.
4. Unclear evidence threshold
A project can drift quickly if the team has not agreed on the difference between a sequence tag, a ranked candidate, and a validation-ready sequence call. That definition belongs in planning, not only in the final report.
Project-Planning Workflow for De Novo Sequencing
Step 1: Define the output class before consuming the sample
Write down what the project must produce. A de novo workflow may generate:
If the decision requires an unambiguous full-length answer, say so at the start. That requirement will shape sample cleanup, acquisition strategy, and validation cost.
A quick comparison helps frame the escalation decision.
| Scenario | Recommended workflow | Main limitation | Next confirmation step |
|---|---|---|---|
| Non-reference sample with strong MS/MS | De novo peptide sequencing first | Some residues may remain ambiguous | Targeted LC-MS/MS or synthetic peptide check |
| Known species with weak spectra | Reacquire LC-MS/MS before de novo work | Interpretation may stay unstable | Repeat run and QC review |
| Mixed fraction with co-fragmentation | Additional purification first | Tags may not support a single candidate | Fraction reassessment |
| PTM-rich peptide with partial mismatch | Combine database search with de novo interpretation | PTM localization may remain uncertain | Site-focused follow-up |
Takeaway: use de novo work when the search space is the main limitation, not when the spectra themselves are the weak point.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
Step 2: Judge whether the sample state supports interpretable output
Define the starting material in practical terms: purified peptide, digest from an intact protein, enriched fraction, gel band, or low-abundance isolate. Sample context changes what can reasonably be recovered.
Purified peptides and cleaner single-band digests are usually better fits for de novo peptide sequencing than broad mixed fractions. For de novo protein sequencing, intact mass context and known processing history can narrow interpretation, even though they do not resolve residue-level ambiguity on their own.
| Sample type | Best-fit objective | Typical constraint | Planning response |
|---|---|---|---|
| Purified peptide | High-confidence candidate reconstruction | I/L ambiguity or PTMs | Reserve material for confirmation |
| Gel band digest | Protein-region reconstruction | Background proteins | Review purity and replicate spectra |
| Enriched fraction | Candidate discovery | Mixed signals | Add purification if full sequence matters |
| Low-abundance isolate | Exploratory sequence tag generation | Sparse fragment evidence | Reassess concentration strategy |
Takeaway: cleaner samples and simpler mixtures make it more likely that the output will support a useful candidate sequence.
Step 3: Set spectrum-centered acceptance criteria
For de novo interpretation, the main raw material is the fragmentation spectrum. A kickoff plan should define what acceptable evidence looks like:
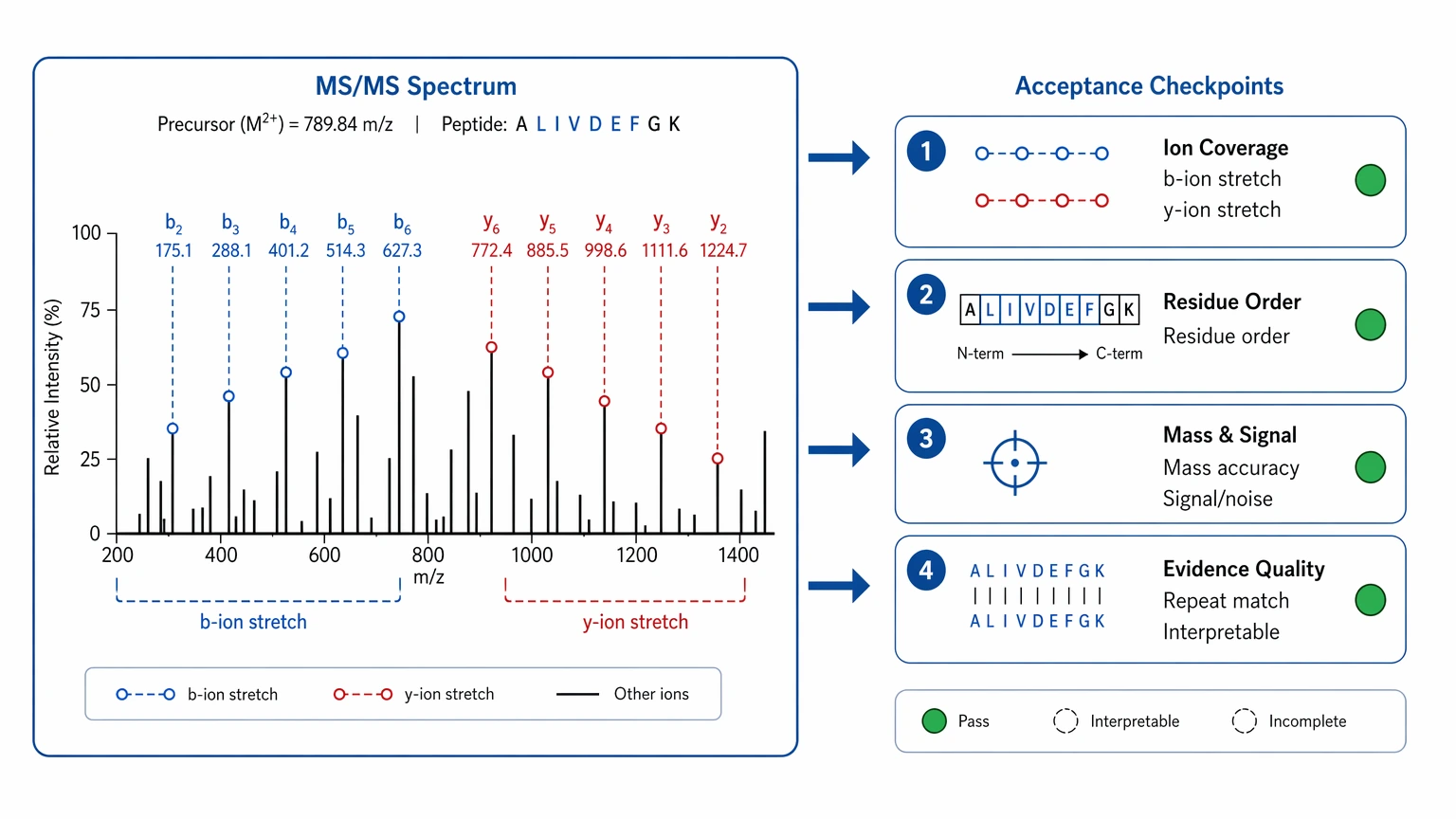
This is where teams need to separate “interpretable” from “complete.” Many useful projects start with a strong sequence tag and then move into targeted confirmation. Trouble starts when a short supported region is treated as if it already proves the entire sequence.
Expected Results and Validation Methods
A well-scoped project usually delivers clearer evidence classification before it delivers full certainty. Immediate deliverables often include:
Follow-up confirmation is a separate step. Orthogonal validation may include:
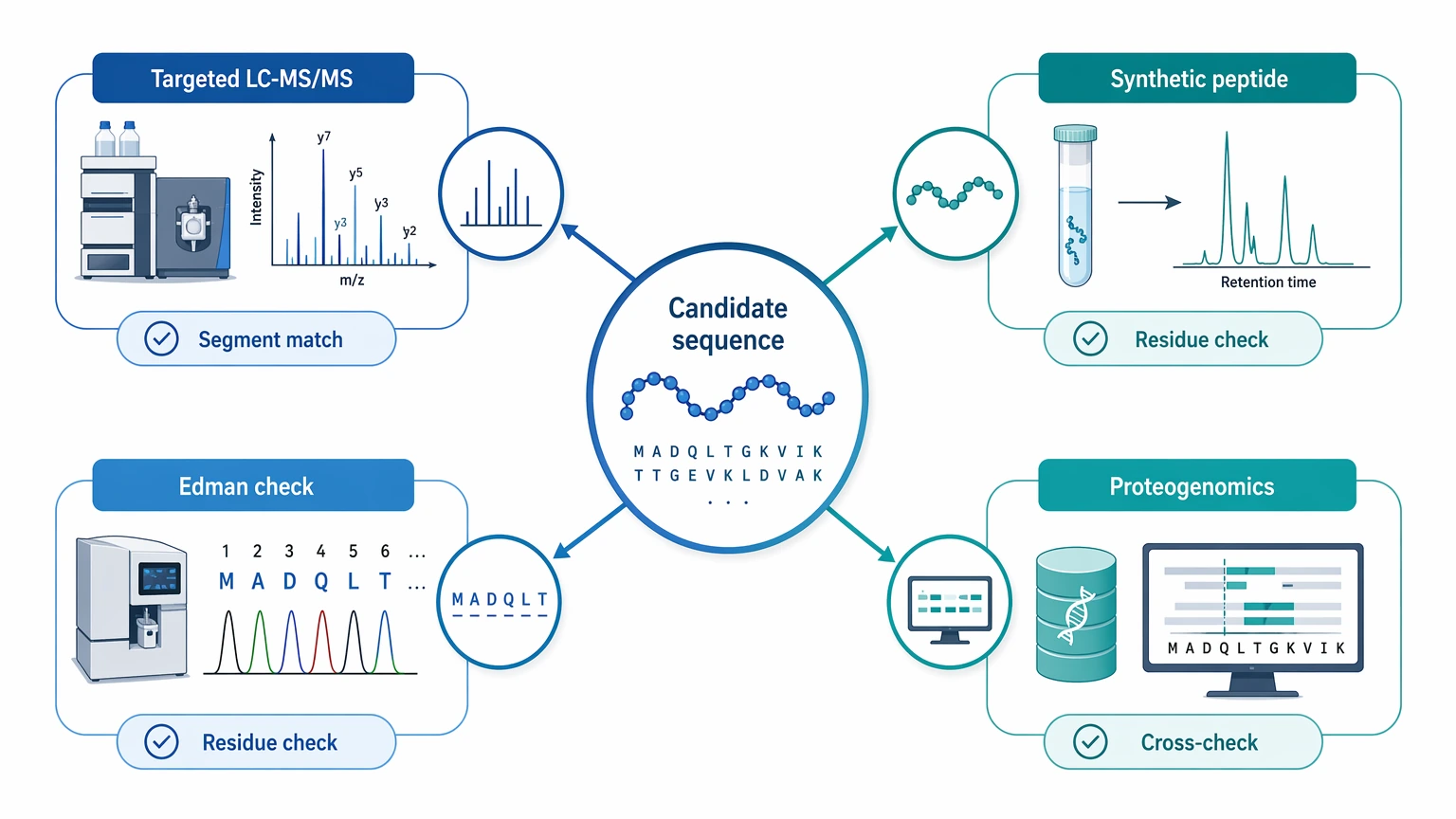
Do not treat a de novo report as self-validating. A candidate supported by strong fragment evidence may be ready for focused confirmation, but publication, synthesis, or functional claims usually need additional support beyond discovery-phase interpretation.
Key Cautions and Practical Limits
Several recurring limits should be built into planning rather than discovered at the end.
Sample quality or amount limits: very low abundance, instability, or severe contamination can reduce usable fragment evidence before interpretation begins.
Controls and repeat expectations: replicate acquisitions help separate stable sequence evidence from one-off assignments. Without repeat support, borderline interpretations carry less weight.
Batch and contamination risk: keratin, carryover, co-isolated precursors, and mixed digest backgrounds can create misleading fragment patterns that look plausible on first review.
Interpretation boundaries: leucine/isoleucine ambiguity and other isobaric residue ambiguity issues may remain unresolved in standard LC-MS/MS data. PTM-driven mass shifts may support a modification hypothesis without fully resolving PTM localization. Database-limited projects can also produce several biologically plausible candidates rather than one final sequence.
When another method is the better next step: if the sample comes from a known organism and the main problem is poor acquisition, incomplete digestion assumptions, or search-parameter setup, a revised database search or cleaner LC-MS/MS run is often the better first move. If full-length certainty is required and the evidence remains fragmented, outside support or a different confirmation method may be the more efficient next step.
What to Ask for Before Approving a Service Scope
Before approving an outsourced project, ask what the report will actually contain. Useful questions include:
If you need help matching sample condition, MS evidence, and expected deliverables, you can submit your requirements to MtoZ Biolabs for project-fit review around de novo peptide sequencing, de novo protein sequencing, and LC-MS/MS report interpretation.
Service Routes to Consider
Conclusion
A de novo LC-MS/MS project is usually justified when standard identification has reached a real reference limit and the sample can still produce interpretable fragment evidence. The strongest plan defines the target output, screens the sample for complexity risk, sets spectrum-based acceptance criteria, and separates immediate candidate deliverables from later confirmation. This framework fits teams working on non-reference samples, modified peptides, novel proteins, and database-mismatched biological material. If your next step is vendor selection or an internal go/no-go review, contact MtoZ Biolabs with the sample type, purification state, LC-MS/MS context, and required decision threshold to evaluate your project against realistic de novo sequencing limits and validation needs.
FAQ
Can de novo sequencing still be useful if the sample contains more than one peptide species?
Yes, but the project goal should usually shift toward candidate discovery or prioritized sequence tags unless purification can reduce mixture complexity first.
Does intact mass information remove sequence ambiguity?
No. Intact mass can constrain interpretation and flag mismatches, but it usually does not resolve residue order or all modification states by itself.
When should a team stop pushing for a single final sequence?
Stop forcing a single answer when several candidates fit the fragment evidence similarly well, or when unresolved I/L positions and PTM uncertainty would still block the downstream decision.
Is database search still worth keeping in the workflow after a de novo project starts?
Often yes. Database search and de novo interpretation are complementary. Sequence tags, modification hypotheses, or narrowed candidate regions can improve the next search round.
What should be ready before the consultation call?
Prepare the sample type, purification state, approximate amount, prior LC-MS/MS results, known biology, intended downstream use, and the minimum confidence level needed for the project decision.
How to order?

