





When Illumina Short Reads Help De Novo Sequencing and When They Become the Assembly Bottleneck
- Keep Illumina as support when peptide-to-ORF mapping is mostly unique and the project needs candidate narrowing or local sequence confirmation.
- Treat Illumina as useful but incomplete when peptide tags map to partial contigs or incomplete ORFs that support only part of the target.
- Escalate beyond short reads when the key question depends on full-length ORF recovery, splice-aware isoform resolution, repeat-rich regions, or discrimination within a homologous protein family.
- Unknown or weakly annotated targets where a standard database search misses the best model
- Novel sequence content where de novo peptide sequencing finds peptide tags that are absent from reference databases
- Local sequence confirmation where the team needs to test whether observed peptide tags fit a predicted coding region from the same sample source
-
Peptide tags map well, but only to partial contigs.
The spectral evidence supports a real sequence segment, but assembly fragmentation prevents full-length protein inference. -
The same peptide-supported region appears in several translated models.
This usually points to paralog ambiguity, weak isoform resolution, or both. -
Predicted ORFs start or stop inside the relevant coding region.
The contig may be real, but the translated product is not complete enough for protein-level interpretation. -
PTM-rich or processed regions sit in weak assembly context.
Strong PTM localization confidence in LC-MS/MS does not, by itself, resolve the surrounding protein architecture. -
Database search failure persists after reasonable database expansion.
That can reflect incomplete assembly-derived databases rather than incorrect residue assignment. - Candidate narrowing: a small set of plausible proteins or ORFs is enough
- Partial reconstruction: confirmed sequence regions plus unresolved regions are acceptable
- Full-length protein inference: the project must distinguish isoforms, termini, repeats, or closely related proteins
- a project-state classification: support, incomplete support, or assembly bottleneck
- a count of peptide tags that map uniquely versus ambiguously
- an assessment of whether mapped regions fall on complete or partial contigs
- a review of ORF completeness around peptide-supported regions
- a statement of whether the current sample-specific database supports local confirmation only or broader protein inference
- For local sequence confirmation: compare predicted translation products with precursor mass and fragment evidence
- For terminal questions: use N- or C-terminal confirmation rather than assuming transcript assembly solved the issue
- For isoform or paralog questions: use orthogonal validation that tests the discriminating region directly
- For PTM-rich targets: confirm sequence architecture and modification assignment as separate but linked questions
- Sample quality or amount limits: low transcript abundance can reduce transcript assembly completeness even when protein enrichment yields strong spectral evidence.
- Controls and repeat expectations: repeat LC-MS/MS runs can stabilize peptide tags, but they do not create missing long-range coding context.
- Batch and contamination risk: mixed material, index carryover, and chimeric assembly can make translation-based annotation look more convincing than it is.
- Interpretation boundaries: peptide-supported local sequence does not by itself prove mature proteoform structure, signal peptide processing, or unique assignment within a homologous protein family.
- When another method is the better next step: if the unresolved question is span-level rather than residue-level, more short-read depth may add local support without resolving the actual decision point.
Short-read Illumina data helps a de novo peptide sequencing or de novo protein sequencing project when it adds sample-specific coding context to strong LC-MS/MS evidence. It becomes the assembly bottleneck when the reporting goal moves from local sequence support to full-length protein inference, isoform resolution, paralog separation, repeat spanning, or terminal definition. When peptide tags keep landing on partial contigs, predicted ORFs do not cleanly cover the peptide-supported region, or several translated models still look plausible, the main constraint is usually no longer tandem mass spectrometry alone. It is short-read-supported reconstruction.
Quick decision block
Where Illumina Short Reads Actually Help
In this workflow, Illumina is not the main answer. It is an evidence layer that strengthens database-independent identification after LC-MS/MS has already produced interpretable peptide tags.
Short reads are most helpful when the target coding region is reasonably expressed, not dominated by long repeats, and not split across several nearly identical transcript models. Under those conditions, transcript assembly and translation-based annotation can produce a sample-specific database that narrows candidate proteins and helps connect spectral evidence to a plausible ORF (open reading frame).
That support matters most in three situations:
This is still a limited claim. Even strong local mapping does not automatically establish a mature proteoform, full terminal structure, or all PTM (post-translational modification) states. A peptide tag can be right while the surrounding protein model is still incomplete.
Signs the Bottleneck Has Shifted from MS/MS to Assembly
Teams often keep refining spectra after the real uncertainty has already moved upstream to transcript assembly or ORF recovery. The most useful distinction here is between local spectral evidence and global coding context.
The bottleneck is likely on the assembly side when you see one or more of these patterns:
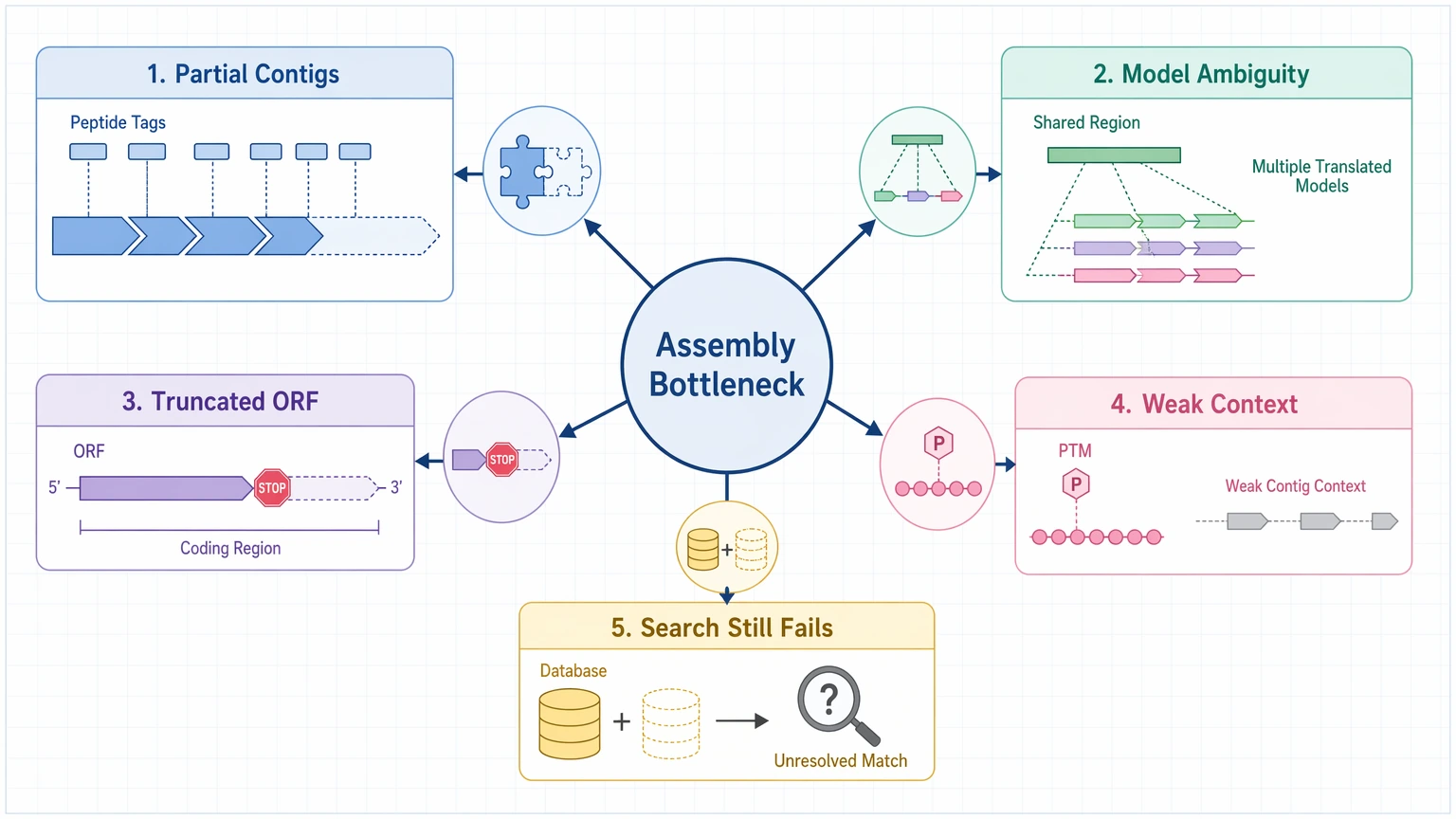
One practical limit should stay in view: when MS/MS coverage is sparse, PTMs are dense, or database-search space is incomplete, sequence confidence may remain local rather than full-length, even if some peptide-to-ORF mapping looks persuasive.
The Most Relevant Root Causes to Check First
For this decision question, four cause categories matter more than a full end-to-end troubleshooting list.
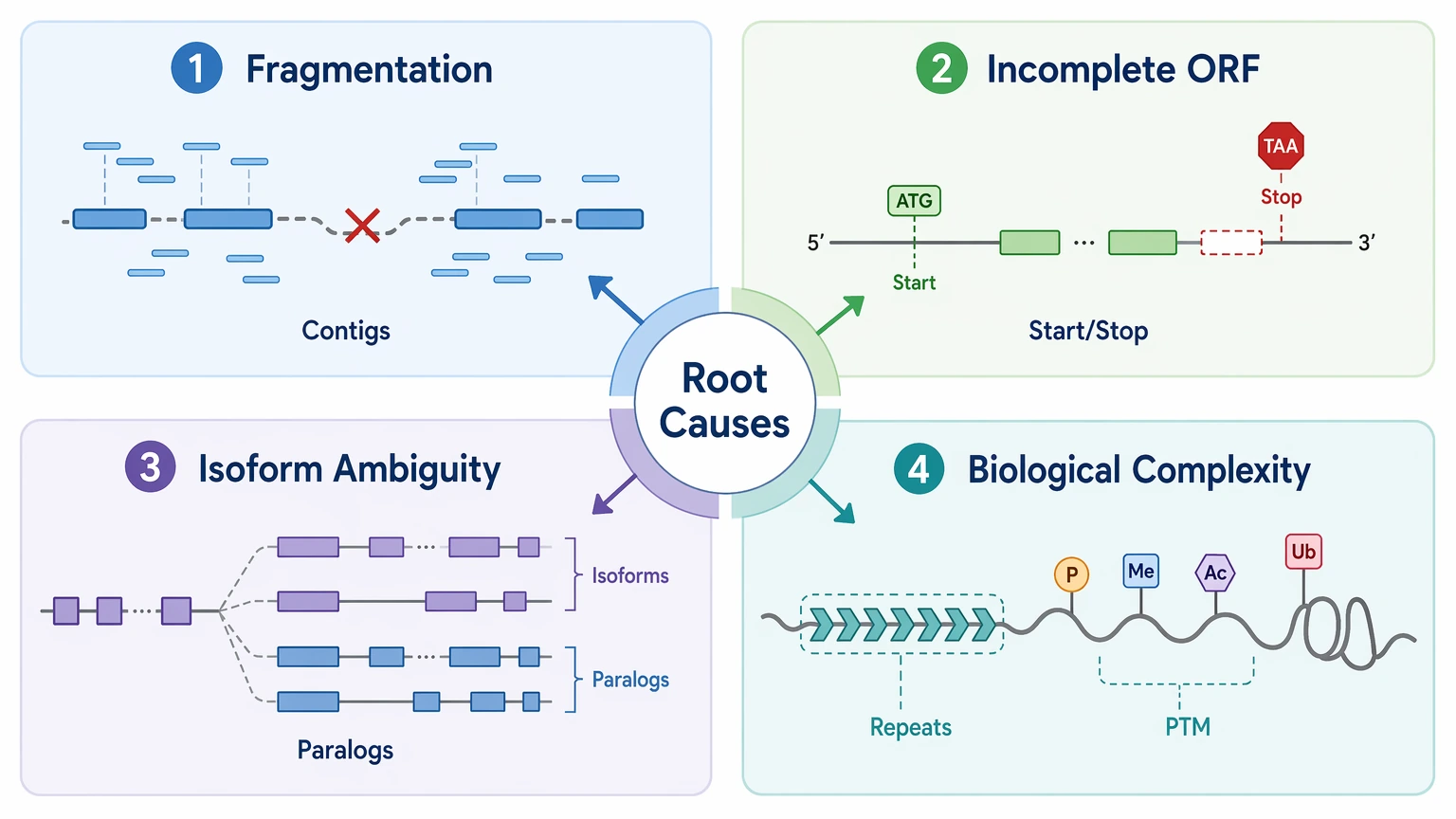
1. Assembly fragmentation
This is the most common reason Illumina support stops short. A contig may contain the peptide-supported region but still fail to connect neighboring exons, terminal regions, or repeated domains.
2. Incomplete ORF recovery
A transcript assembly can look acceptable at the contig level while translation-based annotation still misses the correct start site, stop site, or coding frame. That weakens peptide-to-ORF mapping even when the local sequence evidence is real.
3. Isoform and paralog ambiguity
Short reads can confirm shared regions without separating closely related transcripts or proteins. This is a frequent issue in a homologous protein family and in splice-complex targets.
4. Biological complexity beyond local peptide support
Processed proteins, repeat-rich regions, and PTM-rich proteoforms can all create a gap between strong local residue assignment and uncertain full-length structure.
How to Choose the Right Evidence Strategy
This is a method-selection problem. The goal is not to push every evidence layer equally. The goal is to match the evidence stack to the reporting claim.
Step 1: Define the claim before choosing the workflow
Start with what the final report needs to say.
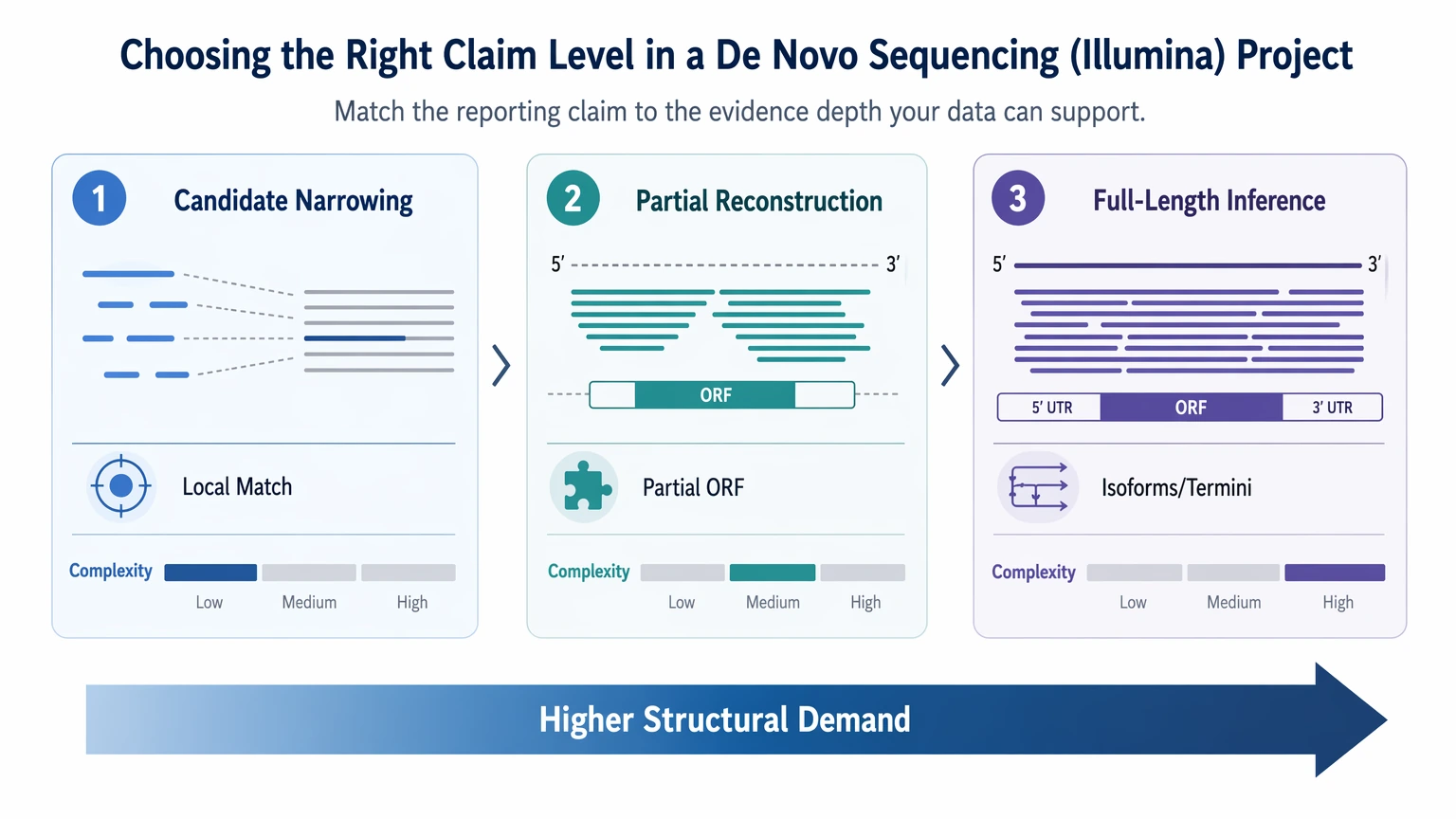
Illumina short reads often fit the first category and can support the second. They are much less dependable for the third when the missing information is structural rather than local.
Step 2: Check whether the Illumina read design fits the reconstruction task
Existing data are not automatically suitable data. Paired-end architecture, insert size, read quality, and depth all shape transcript assembly, especially for low-abundance transcripts. Single-end or short-fragment designs may still help build a sample-specific database, but they are less convincing when the goal is full coding reconstruction.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
The table below helps classify the current project state.
| Scenario | Recommended workflow | Key limitation | Validation need |
|---|---|---|---|
| Peptide tags map uniquely to one ORF region on coherent contigs | Keep Illumina as support | Full termini may still be missing | Check terminal support and mass consistency |
| Peptide tags map to partial contigs with incomplete ORFs | Use Illumina for candidate narrowing | Assembly fragmentation blocks full-length protein inference | Add targeted confirmation |
| Several translated models explain the same peptide set | Treat Illumina support as ambiguous | Isoform resolution or paralog ambiguity | Add orthogonal validation |
| PTM-rich target with strong spectra but weak coding context | Separate PTM interpretation from sequence reconstruction | Local modification evidence does not define the whole proteoform | Validate sequence and modification claims separately |
| Repeat-rich or splice-complex target | Move beyond short-read-only support | Short reads do not span the decisive structure | Add longer-range sequence support or direct protein validation |
Takeaway: this is not a question of whether Illumina is "good" or "bad." It is a question of whether it is being asked to support a local claim or a span-level claim.
Step 3: Decide among three evidence states
State A: Illumina is enough for support
Choose this state when peptide-to-ORF mapping is mostly unique, the relevant ORF is close to complete, and the project goal is confirmation rather than exhaustive structure definition.
State B: Illumina is useful but incomplete
Choose this when the data support part of the sequence model but not enough for a clean protein-level call. This is the most common state in de novo sequencing illumina projects.
State C: Illumina is now the bottleneck
Choose this when the unresolved question depends on structure that short reads do not span well: full-length ORF recovery, isoform resolution, paralog separation, repeat spanning, or terminal definition.
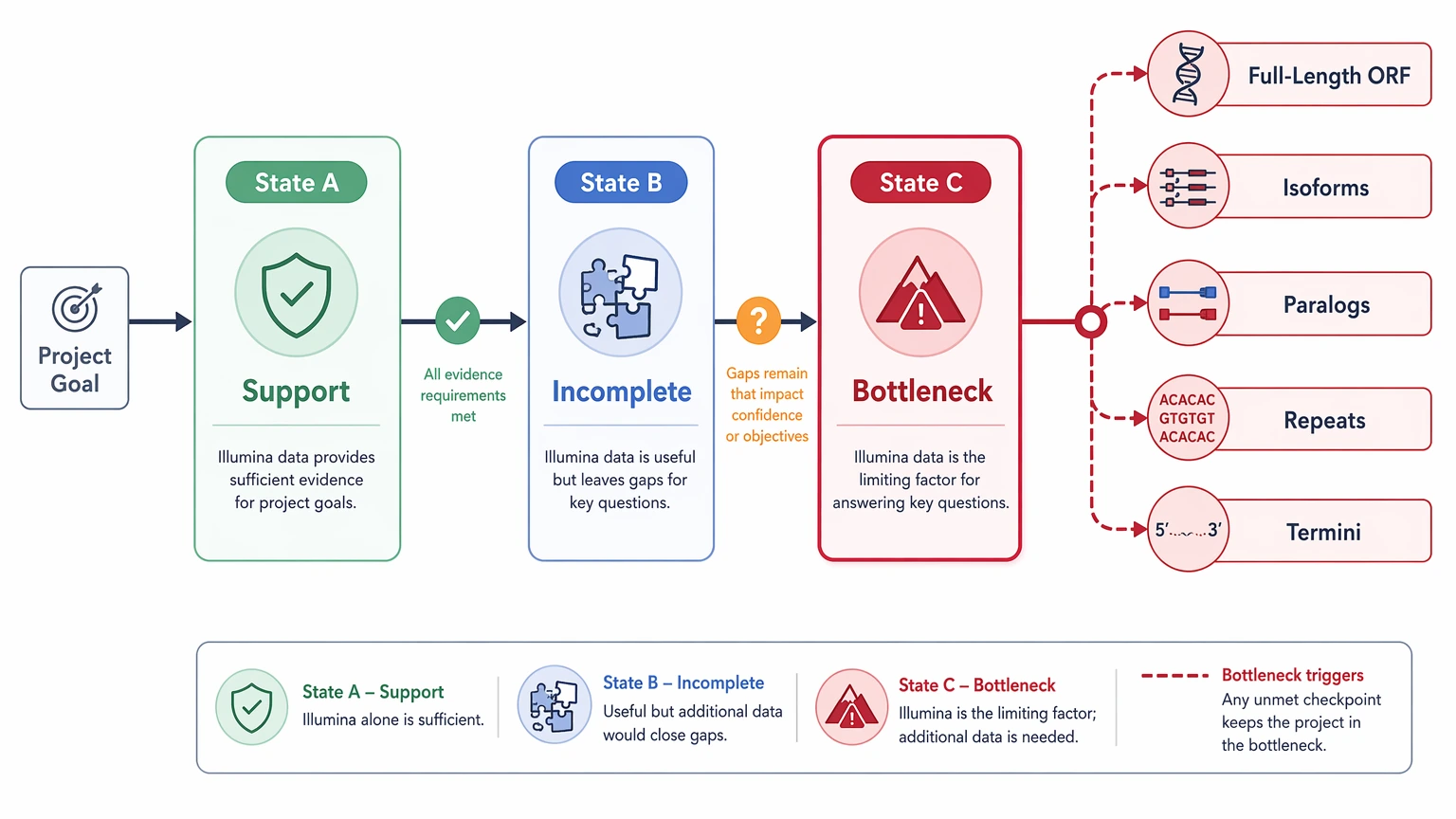
At this transition point, a workflow review is often more useful than another round of generic assembly tuning. If your team needs to separate spectral evidence from assembly limits and decide what to validate next, MtoZ Biolabs can evaluate your project using the LC-MS/MS outputs, assembly files, and reporting goal so you can submit your requirements with a clearer workflow choice.
Expected Results and How to Validate Them
The first expected result is not always a longer sequence. Often, it is a clearer boundary between what is confirmed and what remains uncertain.
Immediate deliverables
After applying this framework, the immediate outputs should include:
These are decision-support deliverables. They show you which layer is limiting.
Follow-up confirmation
Follow-up confirmation depends on the claim you need to defend:
Validation should separate what has already been established from what still needs independent support. A partial contig with matching peptide tags is useful evidence, but it is not the same as a complete full-length protein call.
Key Cautions and Practical Limits
Short-read-supported workflows become misleading when teams ask them to answer a question they are not structurally suited to resolve.
That last case matters most. When the bottleneck is structural, the better next step may be orthogonal validation, a different sequence-support strategy, or outside help with workflow matching rather than another cycle of short-read-only assembly.
Conclusion
Illumina short reads help de novo peptide sequencing and de novo protein sequencing when they strengthen peptide-to-ORF mapping, support a sample-specific database, and narrow plausible coding models around solid LC-MS/MS evidence. They become the assembly bottleneck when the study needs full-length protein inference, strong isoform resolution, paralog separation, repeat spanning, or confident terminal definition. For projects involving unknown proteins, novel isoforms, PTM-rich targets, or fragmented assemblies, the most defensible path is to say plainly whether the current evidence supports confirmation, partial reconstruction, or only a limited local claim. If your project sits in the incomplete or bottleneck category, prepare the spectra, peptide tags, contigs, predicted ORFs, and reporting goal, then contact MtoZ Biolabs to evaluate your project and decide whether short-read support is still fit for purpose or whether a different validation route is the better next step.
FAQ
Can Illumina-derived contigs still be useful if they do not contain a full ORF?
Yes. They can still support local peptide-to-ORF mapping, help build a sample-specific database, and narrow candidates. The limitation is that they do not justify a full-length protein inference on their own.
What is the clearest sign that assembly fragmentation, not poor spectra, is the main problem?
Look for stable peptide tags with credible residue assignment that repeatedly map to edge regions of contigs or to ORFs that change with each assembly model. That pattern points to incomplete coding context rather than weak tandem mass spectrometry.
Should genomic short reads and RNA-seq short reads be interpreted the same way for this use case?
No. RNA-seq data usually connect more directly to transcript assembly and coding reconstruction, while genomic short reads may support sequence context without cleanly resolving expressed isoforms. Their value depends on the biological question being asked.
When does expanding the sample-specific database stop helping?
It stops helping when each new round adds more translated candidates without improving unique peptide support, ORF completeness, or discrimination between competing models. At that point, the database is getting larger, not clearer.
What should a team prepare before requesting workflow review?
Prepare the LC-MS/MS peak files or interpreted spectra, de novo peptide tags, database search summaries, read QC metrics, transcript assembly outputs, predicted ORFs, and a one-sentence statement of the reporting goal.
How to order?

