





Single-Cell Antibody Sequencing Sample Planning: What Cell Input and Sorting Strategy Matter Most for Sequence Recovery
- the target population is a very small fraction of the starting sample
- pre-sort or post-thaw viability is already compromised
- the gate includes mixed B-cell states that do not fit the project goal
- sort purity is uncertain because the phenotype is too broad
- single-cell deposition is planned, but deposition confidence is not checked
- the downstream need is full-length variable-region recovery rather than partial CDR interpretation
- Repertoire survey: broad clonotype structure across a B-cell population
- Rare-clone discovery: recovery of uncommon antigen-specific B cell sequences
- Recombinant re-expression planning: paired heavy/light chain sequences with enough completeness for expression design
- Hybridoma rescue support: sequence recovery from a highly limited but already defined source
- paired heavy/light chain calls only
- near-full heavy chain variable region and light chain variable region reconstruction
- CDR-focused interpretation
- sequence sets suitable for recombinant re-expression
- fraction of cells with interpretable BCR reads
- paired heavy/light chain recovery rate
- usable VH and VL assignment
- full-length variable-region recovery versus partial reads
- clonotype distribution, including duplicate dominance
- consistency between index sorting annotations and recovered sequence groups
- source material: PBMC, enriched B cells, memory B cells, plasmablasts, antigen-specific B cells, or hybridoma-derived cells
- total available starting cells
- estimated target-cell frequency
- pre-sort and post-thaw viability
- expected singlet rate and debris burden
- planned gate and expected sort purity
- whether index sorting will be used
- approximate number of single cells expected for deposition
- whether the endpoint is clonotype survey, rare-clone capture, CDR interpretation, or recombinant re-expression
For most single-cell antibody sequencing projects, the minimum useful input is not the total PBMC count in the tube. It is the number of viable target B cells you can isolate as clean singlets and move into the workflow with real confidence. If the endpoint is antibody sequence recovery, the sorting strategy that usually matters most is the narrowest biologically justified population that still gives you enough single cells for meaningful paired heavy/light chain analysis.
That starting point helps avoid a common planning error: treating total starting cells, sorted cells, and sequence-producing cells as though they are the same number. They are not. A project can start with a large PBMC sample and still yield very few interpretable VH and VL pairs if the target population is rare, viability is weak, or single-cell deposition is inconsistent. On the other hand, a smaller but cleaner antigen-focused sort often supports better full-length variable-region recovery and more usable clonotype interpretation.
Where Sample Planning Usually Breaks Down
Teams often arrive at the sorter with enough material to run something, but not enough clarity to run the right thing. The sample may be total PBMC, enriched B cells, memory B cells, plasmablasts, antigen-specific B cells, or hybridoma-derived single cells. The harder question is usually not whether the platform can capture cells. It is whether the chosen cells are likely to produce interpretable antibody transcripts with native paired heavy/light chain information.
A few warning signs often point to poor antibody sequence recovery:
When those issues stay unresolved, sequencing output often ends up dominated by empty captures, partial heavy chain variable region or light chain variable region reads, repeated expanded clonotypes, or pairs that are not strong enough for recombinant re-expression planning.
The Main Reasons Sequence Recovery Falls Short
A small set of upstream issues explains most failed or underperforming runs.
1. Total cell count is mistaken for target-cell input
A sample can look large on paper and still be a weak fit for the run. If antigen-specific B cell frequency is very low, most of the starting PBMC pool may have little to do with the decision you are trying to make.
2. The sort population does not match the biological question
A broad B-cell receptor (BCR) repertoire survey, a rare binder discovery project, and a plasmablast-focused response study should not start from the same gate. If the population is wrong, sequencing capacity gets consumed without improving the endpoint.
3. Viability, singlet gating, or sort purity is too weak
Cells may pass through the sorter and still fail to contribute usable antibody transcripts. Dead cells, debris, sticky events, and doublets all cut into the fraction of sorted cells that can support interpretable pairing.
4. The required sequence deliverable is stricter than the sample can support
A project that only needs CDR-level interpretation can tolerate more incompleteness than one that needs paired VH and VL sequences suitable for recombinant re-expression.
A Practical Planning Workflow for Better Antibody Sequence Recovery
This article follows a project-planning structure: define the output first, then choose the narrowest sample and sorting path that can realistically support it.
Step 1: Define the real endpoint before deciding how many cells to load
The planning target should be the final sequence use case, not a generic capture count. In practice, most projects fall into one of four categories:
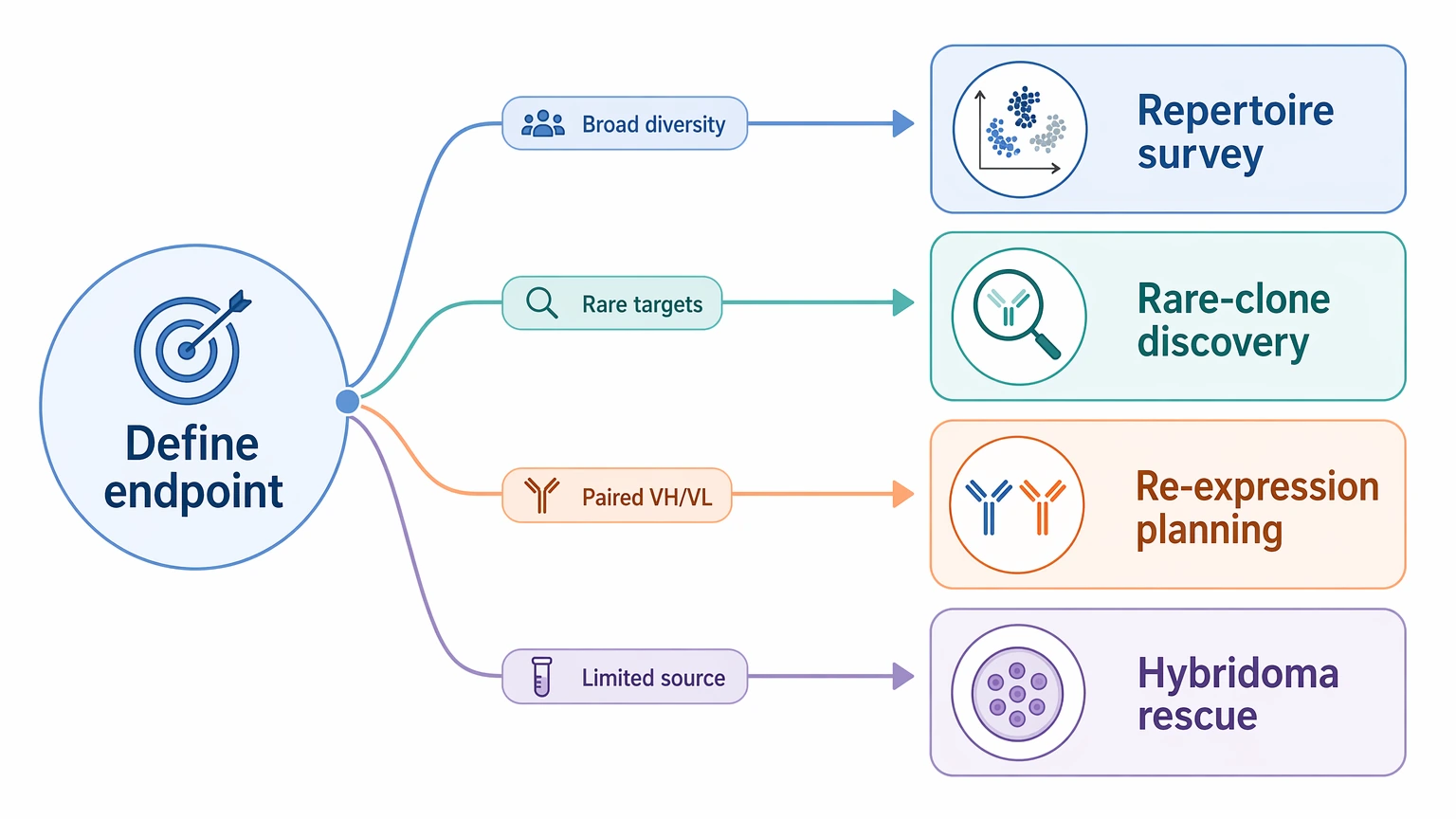
That first decision changes what “enough cells” actually means. A repertoire survey can tolerate a broader gate and some redundancy. Rare-clone discovery usually cannot.
Step 2: Estimate target-cell abundance after real gating losses
The planning number that matters is the estimate after dead-cell exclusion, debris removal, singlet gating, phenotype gating, and handling loss. That figure tells you far more than the starting sample count.
A practical way to think about it is to separate reservoir size from actionable input:
| Planning metric | What to measure | Why it matters |
|---|---|---|
| Total starting cells | Cells available before enrichment or sort | Sets the upper limit, not the likely output |
| Target-cell frequency | Fraction of the intended B-cell subset | Indicates whether enrichment is needed |
| Viability | Pre-sort and post-thaw viable fraction | Affects transcript usability |
| Singlet rate | Fraction passing singlet gating | Flags doublet and deposition risk |
| Sort purity | Purity of the selected population | Improves the density of relevant cells |
| Sorted single cells | Cells actually deposited into the workflow | Best estimate of run value |
| Output requirement | CDR-only versus full-length variable-region recovery | Sets the needed upstream stringency |
If this filtered estimate is small, simply adding more unfractionated cells usually does not fix the real bottleneck.
Step 3: Match the sorted population to the project goal
The sort population should reflect where useful antibody transcripts are most likely to come from for your question.
| Project goal | Sort population that often fits | Main reason |
|---|---|---|
| Broad repertoire sampling | Total B cells or broad memory B cell gate | Preserves diversity for clonotype overview |
| Affinity-matured or recall response profiling | Memory B cell | Enriches for class-switched, experienced cells |
| Acute humoral response studies | Plasmablast | Often increases immunoglobulin transcript abundance |
| Rare binder discovery | Antigen-specific B cell | Concentrates scarce targets |
| Defined rescue scenario | Hybridoma-derived single cells or narrow phenotype gate | Focuses recovery on a known source |
A total B-cell sort is not automatically the wrong choice. It often makes sense for repertoire-oriented work. It becomes inefficient when the real objective is to capture rare binders or connect sequence recovery to a narrow phenotype.
Step 4: Decide whether enrichment is worth the tradeoff
Enrichment adds handling, and extra handling can reduce cell recovery. Even so, it often improves project fit when target-cell frequency is the main constraint. This comes up often with scarce antigen-specific B cells, noisy PBMC backgrounds, or debris-heavy samples where broad loading would spend sequencing capacity on the wrong cells.
Use enrichment when it changes the biological composition enough to improve the expected sequence recovery rate. Skip it when the target population is already abundant and clean enough that direct sorting preserves more viable cells.
Step 5: Add index sorting only when phenotype-to-sequence linkage will change decisions
Index sorting takes more setup, so it should serve a clear downstream purpose. It becomes useful when clone ranking depends on antigen-binding intensity, isotype, maturation state, or marker-defined subpopulations. In those cases, index sorting helps connect each recovered paired heavy/light chain sequence to the original cell event.
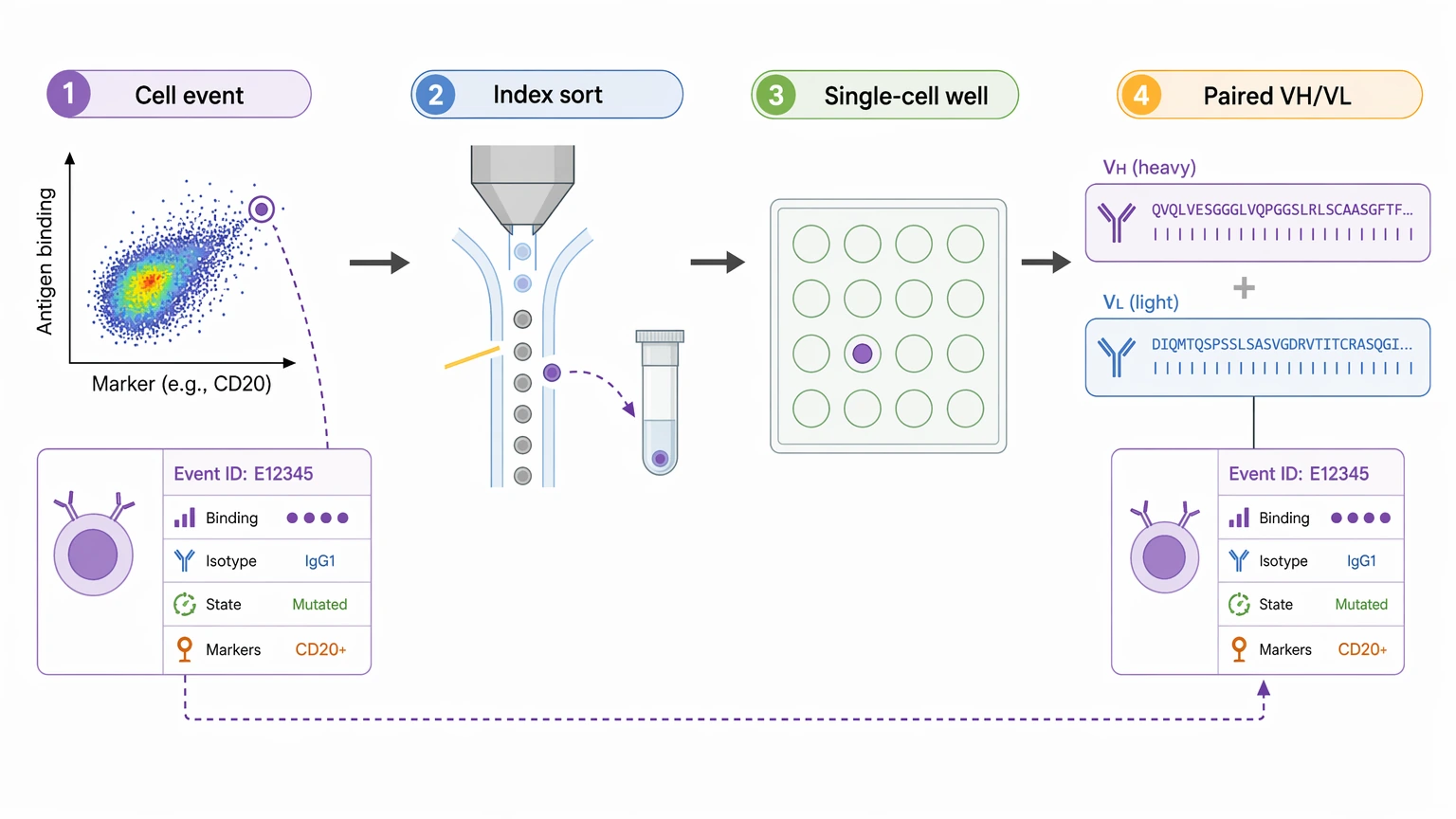
If your team is at that point, you can submit your requirements to MtoZ Biolabs to evaluate whether index sorting, antigen-specific enrichment, or a simpler single-cell deposition workflow is a better match for the sample reality and the desired sequence output.
Step 6: Check whether the sample can support the required sequence format
Not every project needs the same level of sequence completeness. Ask early whether the downstream decision depends on:
This matters because full-length variable-region recovery places tighter demands on viability, sort purity, and single-cell deposition fidelity than exploratory NGS-based screening.
Step 7: Pivot early if the sample is a poor single-cell fit
Some projects should not be pushed into a single-cell antibody sequencing workflow. Consider an alternative when the target population is too rare to enrich credibly, post-thaw viability stays poor, the gated population is biologically uncertain, or the only material available is better suited to hybridoma sequencing or RACE.
That is not a failure of single-cell antibody sequencing. It is a decision about fit. A narrower question with a different recovery route can produce more interpretable antibody sequence recovery than a nominal single-cell run built on weak starting assumptions.
What a Stronger Plan Should Improve
A stronger plan does not promise a fixed sequence recovery rate, but it should raise the fraction of sorted cells that return useful antibody information.
After execution, focus on checks that matter to the project:
These checks tell you more than cell capture counts by themselves. If the output is dominated by partial calls or a few repeated clonotypes, the next review should begin with target-cell definition, sort purity, and deposition quality rather than simply scaling up total input.
A Pre-Consultation Checklist for Single-Cell Run Planning
Before requesting project review, organize the sample facts that actually affect antibody sequence recovery:
With that information in hand, contact us or evaluate your project with MtoZ Biolabs so the team can compare single-cell antibody sequencing against hybridoma sequencing or RACE when sample limitations make the original plan inefficient.
Conclusion
In single-cell antibody sequencing, the planning number that matters most is the count of viable target cells that can be sorted as clean single cells, not the bulk sample count listed at intake. The sort strategy most likely to improve antibody sequence recovery is the one that stays tightly aligned with the biological goal, preserves viability, supports strong singlet gating, and matches the required level of VH and VL completeness. This approach is especially useful for PBMC-derived antibody discovery, rare antigen-specific B cell capture, plasmablast-focused studies, and rescue-oriented projects where paired heavy/light chain recovery is the true endpoint. If you are deciding between a broader sort, a narrower enriched gate, or an alternative route such as hybridoma sequencing or RACE, prepare the target-cell estimate, sample-quality data, and downstream sequence goal first, then contact us for a focused planning discussion.
FAQ
If my PBMC sample is large, can I skip target-cell frequency estimates?
Not safely. A large PBMC pool can still leave you with too few relevant B cells after singlet gating and phenotype selection to support useful paired heavy/light chain analysis.
Are naive B cells ever useful in single-cell antibody sequencing?
They can be, but they are usually a weaker starting point for discovery-driven projects that prioritize affinity-matured or antigen-experienced antibodies. If your goal is rare binder recovery, a memory B cell or antigen-specific B cell strategy is often more informative.
Does a higher sorted cell count always improve clonotype diversity?
No. A broader sort can raise cell count while also bringing in more biologically irrelevant cells or duplicate expanded clones. Diversity capture depends on the composition of the sorted population, not just on how many cells were loaded.
When should I lower the ambition from full-length variable-region recovery to CDR-focused interpretation?
Consider that shift when sample integrity is borderline, target-cell numbers are limited, or the project question can be answered without near-complete VH and VL reconstruction. A narrower deliverable is usually better than assuming expression-ready sequence pairs from weak input.
Can index sorting replace sort purity?
No. Index sorting adds metadata, but it does not repair a poorly defined gate or a mixed-cell deposition problem. You still need strong sort purity and reliable single-cell deposition for interpretable pairing.
What information helps a service team judge whether RACE or hybridoma sequencing is a better alternative?
The key details are sample source, whether live sortable single cells are available, target-cell frequency, viability, and whether the project needs native heavy/light pairing or simply sequence recovery from a defined antibody-producing source.
How to order?

