





Protein Sequencing: Principles, Workflows, and Research Applications
Introduction
Protein sequencing determines the amino acid order of a protein or peptide. This information supports cloning, expression design, antibody engineering, biopharmaceutical characterization, and publication of primary structure evidence. In many projects, researchers already know that a protein is present, but the exact sequence remains uncertain. A purified band on a gel, a recombinant batch with incomplete records, or an antibody with no genetic source can all create this gap. Without reliable sequence data, downstream decisions about construct design, QC release, and functional validation become harder to defend.
Modern protein sequencing combines classical chemistry with high-resolution mass spectrometry. Edman degradation reads amino acids stepwise from the N-terminus. LC-MS/MS methods digest proteins into peptides, interpret fragmentation spectra, and assemble sequence evidence at peptide or protein level. Database-assisted identification works when a correct reference exists. De novo sequencing works when the reference is missing, incomplete, or untrustworthy. The right workflow depends on sample type, sequence coverage needed, modification status, and whether the goal is terminal confirmation or full-length recovery.
Related Services
|
Service Area |
Recommended Service |
|---|---|
|
De novo protein sequencing |
|
|
Full-length protein sequencing |
|
|
Terminal sequencing |
N-Terminal Sequencing Service / C-Terminal Sequencing Service |
|
Classical sequencing |
|
|
Antibody sequencing |
Researchers evaluating protein sequencing options can consult MtoZ Biolabs to match sample type, coverage goal, and reporting format before sample submission.
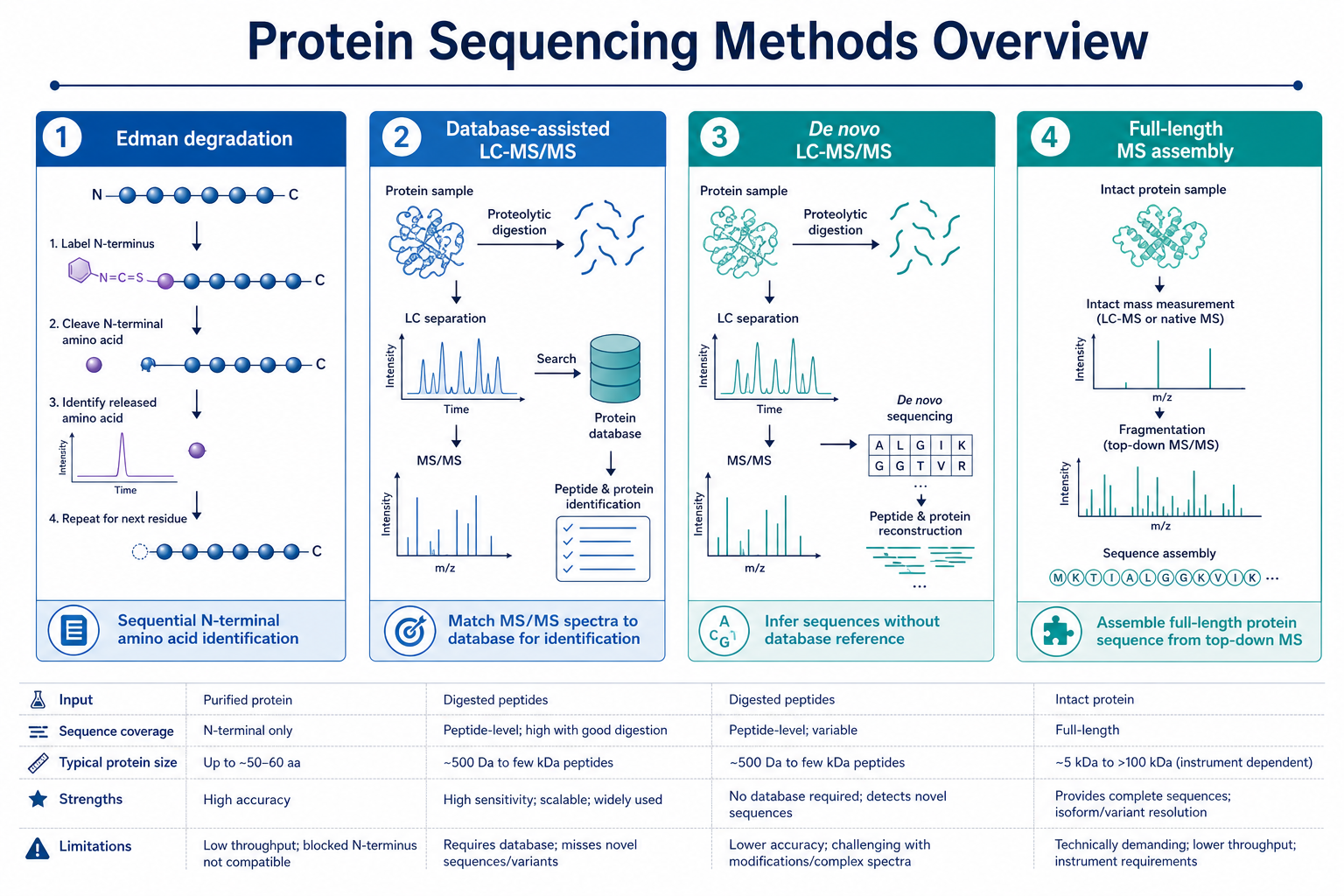
Figure 1. Protein sequencing methods range from terminal Edman degradation to database search, de novo MS sequencing, and full-length assembly.
What Protein Sequencing Means in Practice
Protein sequencing answers a practical question: what is the amino acid sequence of this protein sample? The answer may be partial or complete. A terminal sequencing result may confirm the first 10 to 30 residues from the N-terminus or C-terminus. A peptide-mapping result may confirm that an expressed product matches an expected reference. A de novo sequencing result may reconstruct a sequence when no reliable reference exists.
The method choice depends on the information needed. Terminal sequencing is efficient when only the start or end of the protein must be verified. Database-assisted LC-MS/MS is efficient when a high-quality reference is available and the goal is confirmation. De novo sequencing is needed when the protein is unknown, proprietary, truncated, or poorly represented in databases. Full-length sequencing often combines multiple protease digests, overlapping peptide evidence, and expert sequence assembly.
Core Principles of Major Sequencing Approaches
1. Edman Degradation
Edman degradation removes one N-terminal amino acid per cycle and identifies each released residue. The method is direct, well established, and useful for N-terminal confirmation of purified proteins and peptides. It is less suited to long full-length sequencing because signal loss accumulates over many cycles. Modified N-termini, blocked termini, and low sample amount can also limit performance.
2. Database-Assisted LC-MS/MS Identification
In this workflow, a purified protein is digested with a protease such as trypsin. Peptides are separated by liquid chromatography and analyzed by tandem mass spectrometry. Peptide-spectrum matches are made against a protein database. When the correct reference exists and spectral quality is strong, this approach can confirm identity and map coverage across the sequence.
The limitation is database dependence. If the sample contains an unexpected variant, contamination, or a sequence absent from the database, the method may return a partial match or an incorrect assignment.
3. De Novo LC-MS/MS Sequencing
De novo sequencing interprets peptide fragmentation patterns without relying on a prior database match. Analysts derive sequence tags from b-ions and y-ions in MS/MS spectra. Overlapping peptides from one or more digests are assembled into longer regions and, when coverage allows, a full protein sequence.
This approach is central to unknown protein characterization, legacy sample recovery, and recombinant product verification when genetic records are incomplete. Homologous sequences, isobaric residues, post-translational modifications, and incomplete digestion can still create ambiguity, so expert review remains important.
4. Full-Length Sequence Assembly
Full-length protein sequencing usually requires broad peptide coverage across the protein backbone. Multiple proteases, repeat LC-MS/MS runs, and orthogonal terminal confirmation can improve confidence. Intact mass measurement, N-terminal sequencing, and C-terminal sequencing can support the assembled sequence by confirming termini, truncations, or unexpected processing events.
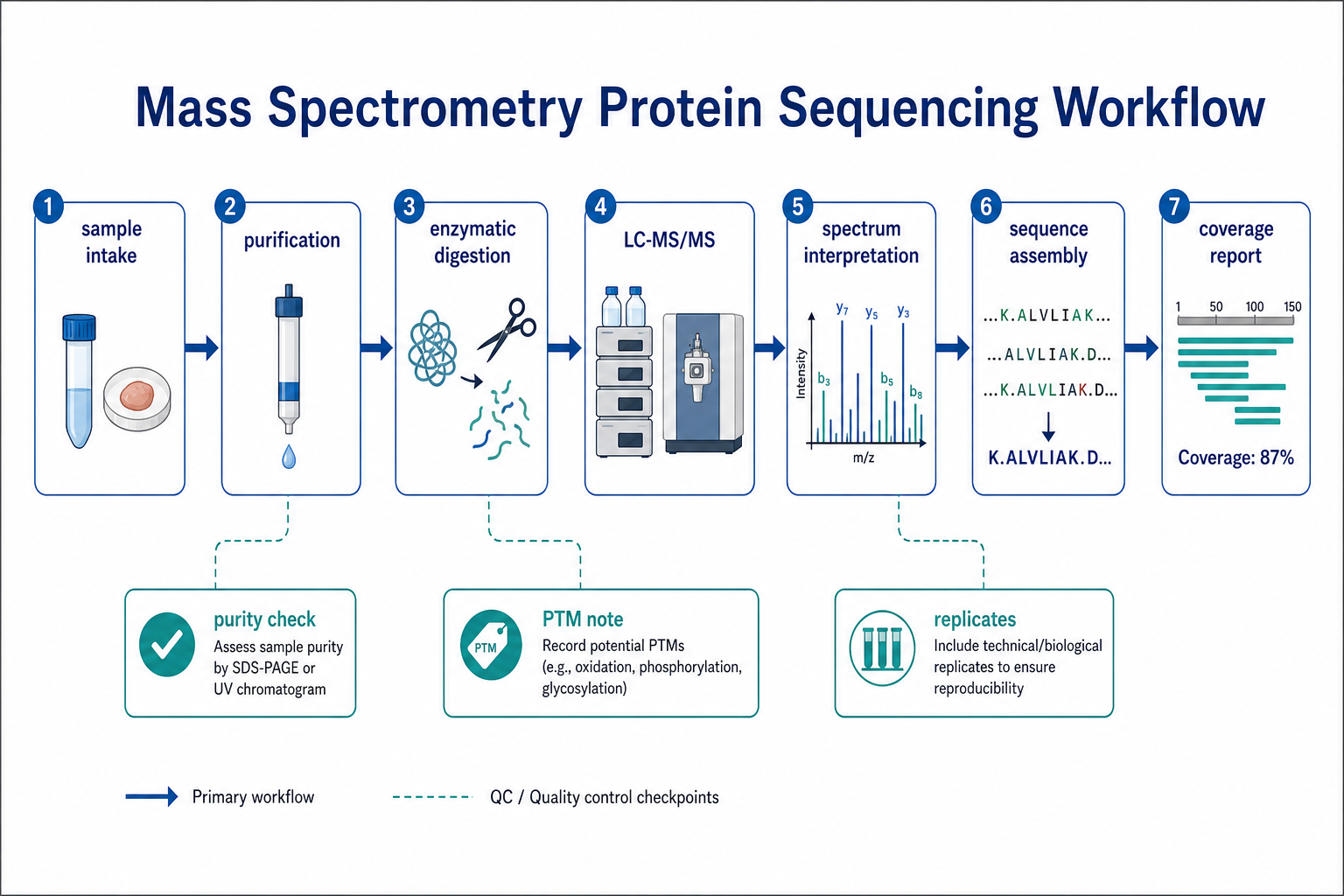
Figure 2. A typical LC-MS/MS protein sequencing workflow includes purification, digestion, spectral interpretation, sequence assembly, and coverage reporting.
Standard LC-MS/MS Protein Sequencing Workflow
A robust protein sequencing project usually follows a defined sequence of steps. Each step affects coverage, confidence, and report usability.
1. Sample Feasibility Review
Purity, amount, buffer composition, and expected protein length are assessed before digestion.
2. Sample Preparation
Proteins may be purified further, reduced, alkylated, or separated by gel band excision when needed.
3. Enzymatic Digestion
Trypsin is common, but alternative proteases can improve coverage across difficult regions.
4. LC-MS/MS Acquisition
High-resolution tandem mass spectrometry generates peptide fragmentation data.
5. Spectrum Interpretation
Peptides are identified by database search, de novo analysis, or both.
6. Sequence Assembly
Overlapping peptides are aligned into contiguous sequence regions.
7. Coverage Mapping and QC Review
Gaps, modifications, and low-confidence regions are documented in the final report.
Sample purity strongly affects outcome. A dominant protein band can support high-confidence sequencing. A complex mixture may require additional fractionation before meaningful sequence assembly is possible. For antibody or biopharmaceutical samples, matrix interference and glycosylation should be considered during project design.
Method Comparison
|
Method |
Best Use Case |
Main Strength |
Main Limitation |
|---|---|---|---|
|
Edman degradation |
N-terminal confirmation |
Direct residue readout from the N-terminus |
Low efficiency for long full-length sequencing |
|
Database-assisted LC-MS/MS |
Known or reference-backed proteins |
Fast confirmation when reference quality is high |
Depends on correct database entry |
|
De novo LC-MS/MS |
Unknown or proprietary proteins |
Works without a trusted reference |
Requires strong spectra and expert assembly |
|
Full-length MS assembly |
Complete primary structure documentation |
Broad coverage across the protein |
Higher sample and analysis demand |
|
Antibody sequencing |
Immunoglobulin variable regions |
Supports antibody engineering and recovery |
Higher complexity due to sequence variability |
This comparison shows why protein sequencing is not a single assay. It is a family of methods selected by the biological question and the quality of existing sequence information.
Core Advantages and Current Limitations
1. Core Advantages
(1) Primary structure evidence from protein material. Protein sequencing provides direct evidence from purified protein, which is valuable when genetic information is unavailable.
(2) Support for unknown and proprietary samples. De novo sequencing can recover sequence information when database search is not sufficient.
(3) Compatibility with biopharmaceutical workflows. Terminal sequencing, peptide mapping, and full-length assembly can support QC, comparability, and regulatory documentation.
(4) Orthogonal confirmation options. MS-based sequencing can be combined with Edman degradation, intact mass analysis, and peptide mapping for stronger evidence chains.
2. Current Limitations
(1) Coverage depends on sample quality. Low purity, insufficient amount, or harsh storage conditions can reduce peptide recovery and sequence confidence.
(2) Modifications add complexity. Phosphorylation, glycosylation, oxidation, and other post-translational modifications can complicate interpretation.
(3) Isobaric residues create ambiguity. Leucine and isoleucine cannot always be distinguished by mass alone in routine workflows.
(4) Full-length sequencing is not automatic. Long proteins, repetitive regions, and low-abundance proteoforms may require additional runs or complementary methods.
Researchers should define the required evidence level before starting. A terminal check, a partial coverage map, and a full-length sequence report answer different project needs.
Typical Research Applications
Protein sequencing supports a wide range of research and development workflows.
1. Unknown Protein Identification
A band from gel electrophoresis, an enriched fraction, or a novel purified protein may require de novo sequence recovery before cloning or functional studies can continue.
2. Recombinant Product Verification
Expression batches can be checked against intended design to detect truncation, sequence drift, or incorrect construct expression.
3. Antibody Sequence Recovery
Hybridoma-derived or purified antibody material may require variable-region sequencing for engineering, backup, or redevelopment when genetic material is limited.
4. Biopharmaceutical Primary Structure Analysis
Terminal sequencing, peptide mapping, and modification-aware LC-MS/MS support release testing, comparability, and structure confirmation.
5. Proteoform and Processing Analysis
N-terminal processing, signal peptide removal, C-terminal heterogeneity, and other proteoforms can be documented with the right combination of terminal and peptide-level sequencing.
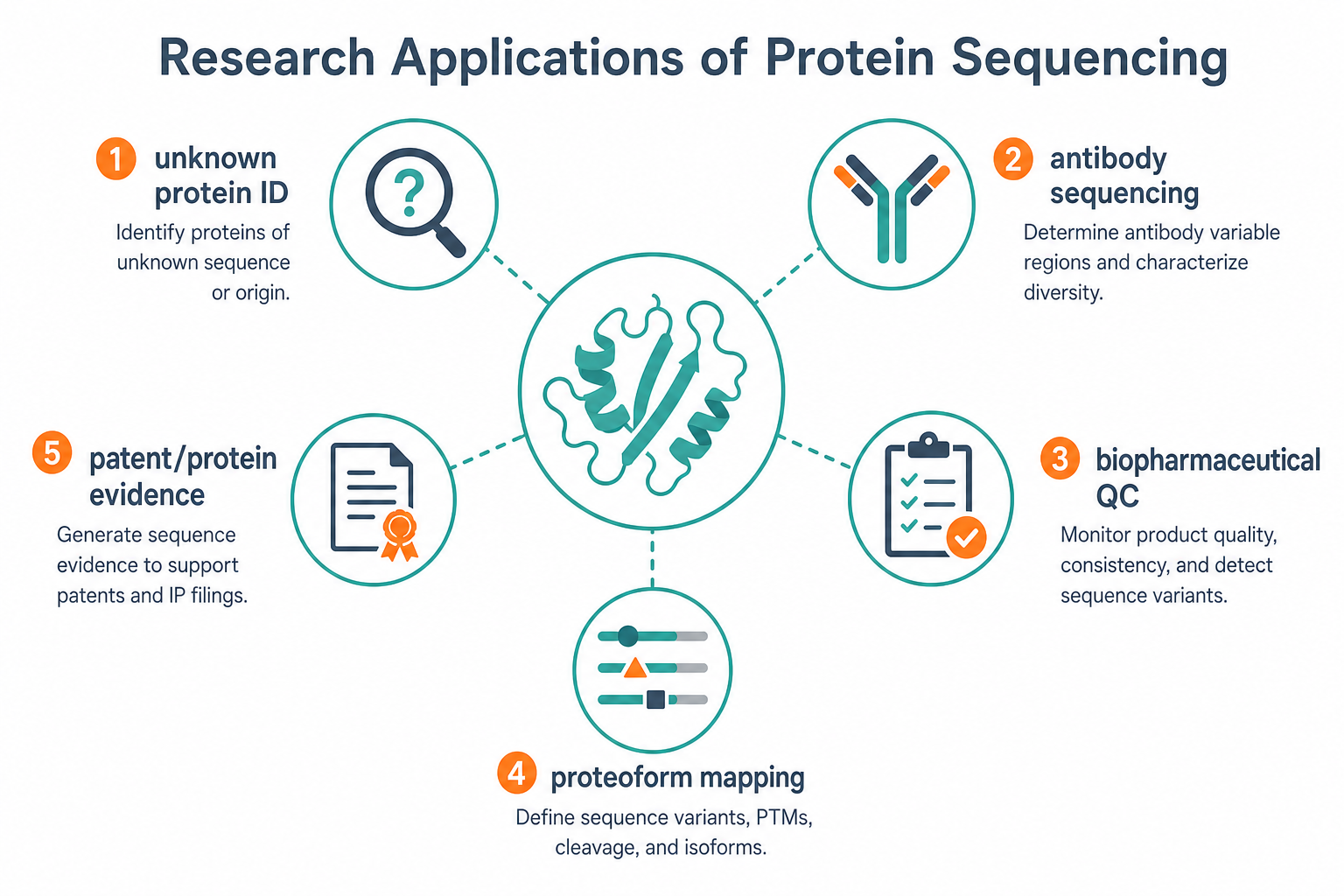
Figure 3. Protein sequencing supports unknown protein identification, antibody sequencing, biopharmaceutical QC, proteoform mapping, and primary structure documentation.
How to Choose the Right Sequencing Strategy
The best strategy depends on three factors: the existing sequence information, the coverage required, and the sample constraints.
If the protein has a trusted reference and the goal is confirmation, database-assisted peptide mapping or protein identification may be sufficient. If only the N-terminus or C-terminus must be verified, Edman degradation or targeted terminal MS methods may be the most efficient choice. If the sequence is unknown or unreliable, de novo LC-MS/MS sequencing or full-length assembly should be considered.
Sample amount also matters. Terminal sequencing can work with relatively small amounts of purified material. Full-length sequencing usually benefits from more starting protein and cleaner preparation. For antibodies, variable-region complexity and glycosylation should be discussed during project planning.
A practical decision path is to define the final deliverable first. Does the project need a terminal check, a coverage map, or a complete sequence report? That decision determines method selection, digestion design, and whether orthogonal confirmation is required.
Future Outlook
Protein sequencing continues to move toward higher sensitivity, better modification handling, and more automated de novo interpretation. Improvements in mass spectrometry acquisition, chromatographic separation, and sequence assembly software are expanding the range of samples that can support reliable primary structure recovery. At the same time, expert review remains important because sample complexity, homology, and modifications still require project-specific judgment.
For many laboratories, the most efficient path is not to build every sequencing capability internally. Outsourcing can provide access to specialized digestion strategies, high-resolution LC-MS/MS, and reporting formats suited to publication or QC needs. The key is to match the service scope to the evidence level required by the project.
Frequently Asked Questions
1. What is the difference between protein identification and protein sequencing?
Protein identification usually matches peptides to a database entry and confirms identity against a known reference. Protein sequencing goes further by determining amino acid order, especially when the reference is missing, incomplete, or needs direct confirmation from the sample.
2. When is de novo protein sequencing needed?
De novo protein sequencing is needed when no reliable reference sequence exists, when the sample may contain an unexpected variant, or when the project requires primary structure evidence independent of database search.
3. Can LC-MS/MS determine a full protein sequence?
Yes, when peptide coverage is sufficient. Full-length sequencing often requires multiple protease digests, overlapping peptides, and expert assembly. Terminal confirmation may be added for higher confidence.
4. Is Edman degradation still useful?
Yes. Edman degradation remains valuable for N-terminal confirmation, especially for purified proteins and peptides when only terminal information is required.
5. How much sample is needed for protein sequencing?
Sample requirements depend on protein length, purity, and method. Terminal sequencing may work with microgram-level purified material. Full-length LC-MS/MS sequencing generally benefits from more input and cleaner preparation. A feasibility review before submission is recommended.
Conclusion
Protein sequencing provides the primary structure evidence needed for discovery, engineering, and quality control. Edman degradation supports direct N-terminal analysis. Database-assisted LC-MS/MS confirms proteins with reliable references. De novo sequencing and full-length assembly recover sequence information when the reference is uncertain. The strongest project outcomes come from matching the workflow to sample quality, coverage needs, and the level of proof required. For unknown proteins, antibody materials, or biopharmaceutical sequence confirmation, researchers can contact MtoZ Biolabs to review sample suitability and select a protein sequencing workflow aligned with the project goal.
How to order?

