





Protein Analysis Mechanisms: How Extraction, Separation, Identification, and Quantification Work
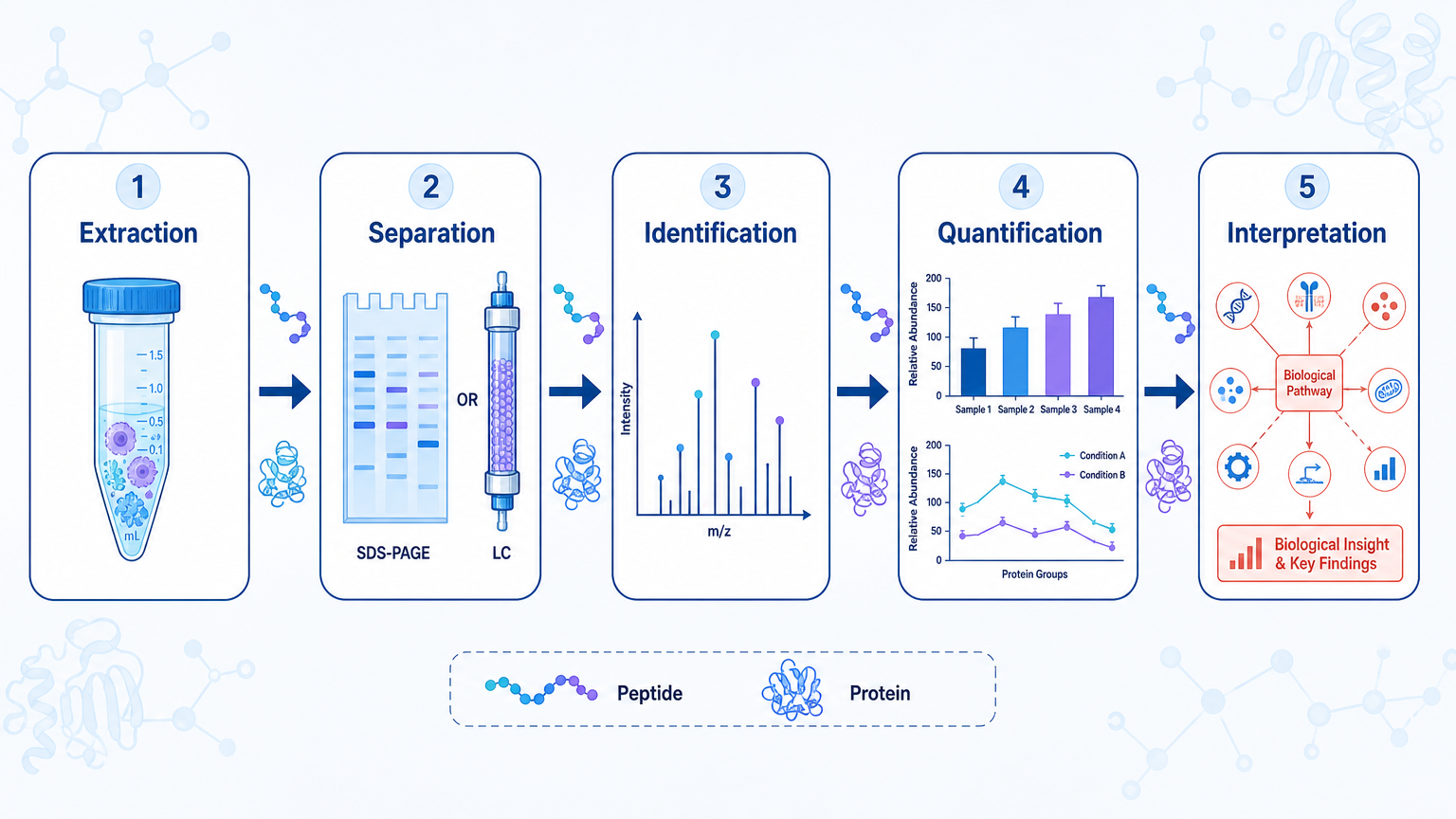
- Protein analysis usually moves through five mechanisms: extraction, separation, detection, identification, and quantification.
- LC-MS/MS is central for broad protein identification, PTM characterization, and proteomics-scale quantification.
- Antibody-based methods such as ELISA and Western blot are useful when the target protein is already known.
- PTM analysis often needs enrichment or targeted acquisition because modified peptides are usually low-abundance.
- Data interpretation depends on sample quality, protein coverage, peptide evidence, controls, and validation strategy.
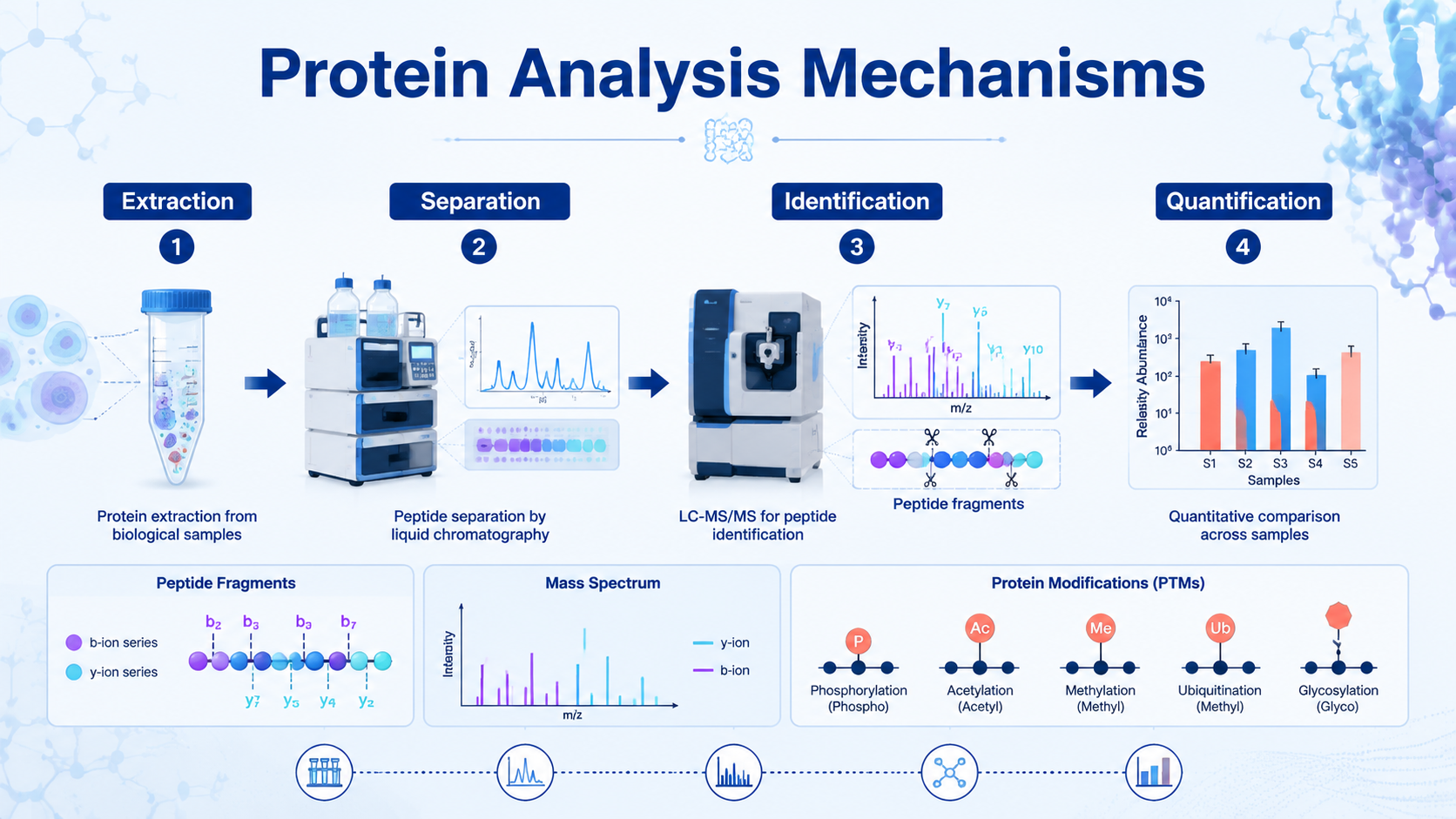
Protein analysis is the set of experimental and computational steps used to extract proteins from biological samples, separate complex mixtures, identify protein species, measure abundance, and characterize structure or modifications. The mechanism is not one technique. It is a workflow: sample preparation creates measurable proteins or peptides, analytical separation reduces complexity, mass spectrometry or antibody-based detection generates signals, and bioinformatics turns those signals into biological interpretation.
Key Takeaways
What Does Protein Analysis Mean in Practice?
Protein analysis answers different questions depending on the project. A biologist may ask whether a protein is present. A biopharma team may need intact mass, purity, sequence coverage, glycosylation status, or host cell protein risk. A disease researcher may need differentially expressed proteins, pathway changes, or candidate biomarkers.
These goals use overlapping methods, but the workflow should be chosen from the question backward. Asking "what protein is in this gel band?" is different from asking "which proteins change across disease groups?" or "which phosphorylation sites respond to treatment?"
Related Services
LC-MS Protein Analysis Service
Mass Spec Protein Analysis Service
Protein Identification by LC-MS/MS Service
Intact Protein Analysis Service
LC-MS Protein Quantification Service
Mechanism 1: Protein Extraction and Sample Preparation
Protein analysis begins by releasing proteins from cells, tissues, biofluids, gels, or formulated products while preserving the features that matter for the study. Lysis buffers disrupt membranes and solubilize proteins. Protease and phosphatase inhibitors may be needed when degradation or dephosphorylation would change the result.
The sample matrix determines the preparation strategy. Tissue may need mechanical homogenization. Plasma may require dynamic range management. Gel bands require in-gel digestion. Biopharmaceutical samples may need buffer exchange, denaturation control, reduction, alkylation, or deglycosylation depending on the question.
Poor preparation creates problems that no downstream software can fully fix: protein loss, incomplete digestion, detergent carryover, salt contamination, oxidation artifacts, and biased recovery of membrane or low-abundance proteins.
Mechanism 2: Protein Separation
Separation reduces complexity before detection. SDS-PAGE separates proteins mainly by size. Two-dimensional PAGE separates by isoelectric point and molecular weight. Liquid chromatography separates proteins or peptides by hydrophobicity, charge, size, affinity, or other physicochemical properties.
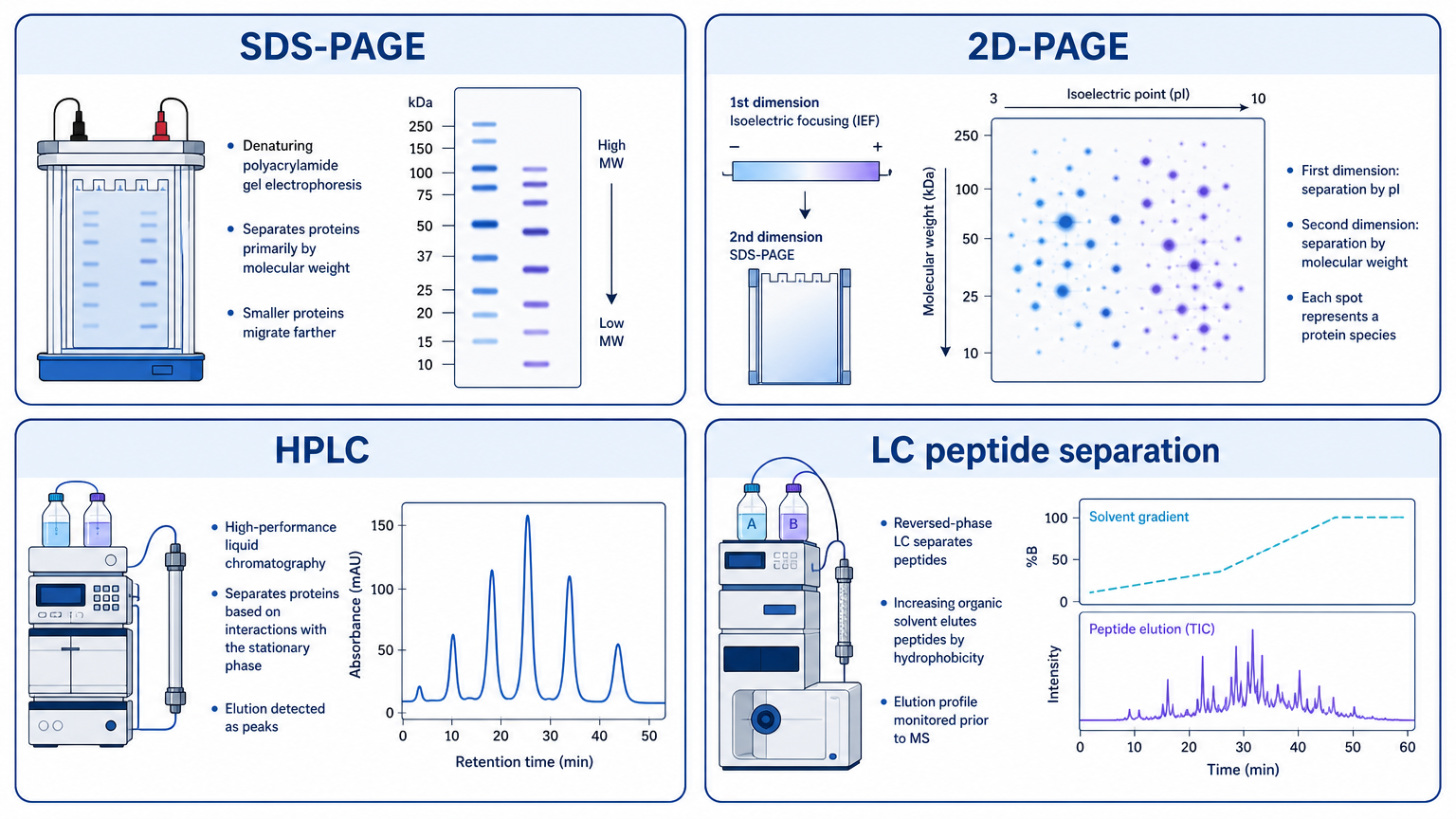
In proteomics, LC separation before MS/MS is especially important because biological samples can contain thousands of proteins and many more peptides. Better separation reduces ion suppression, improves peptide detection, and increases the chance of identifying low-abundance species.
Mechanism 3: Detection and Identification by Mass Spectrometry
Mass spectrometry identifies proteins by measuring peptide ions and matching fragmentation spectra to peptide sequences. In bottom-up proteomics, proteins are digested into peptides, peptides are separated by LC, ionized by ESI or MALDI, measured by mass analyzer, fragmented, and interpreted by database search.
The core MS mechanism includes ionization, mass analysis, fragmentation, detection, and database matching. Protein identification is strongest when supported by multiple unique peptides, good fragmentation spectra, appropriate false discovery rate control, and sample-specific database selection.
Mechanism 4: Protein Quantification
Protein quantification measures abundance. Broad proteomics can use label-free quantification, DIA, TMT, iTRAQ, or SILAC. Targeted quantification can use PRM, SRM/MRM, ELISA, or Western blot depending on specificity, throughput, and validation needs.
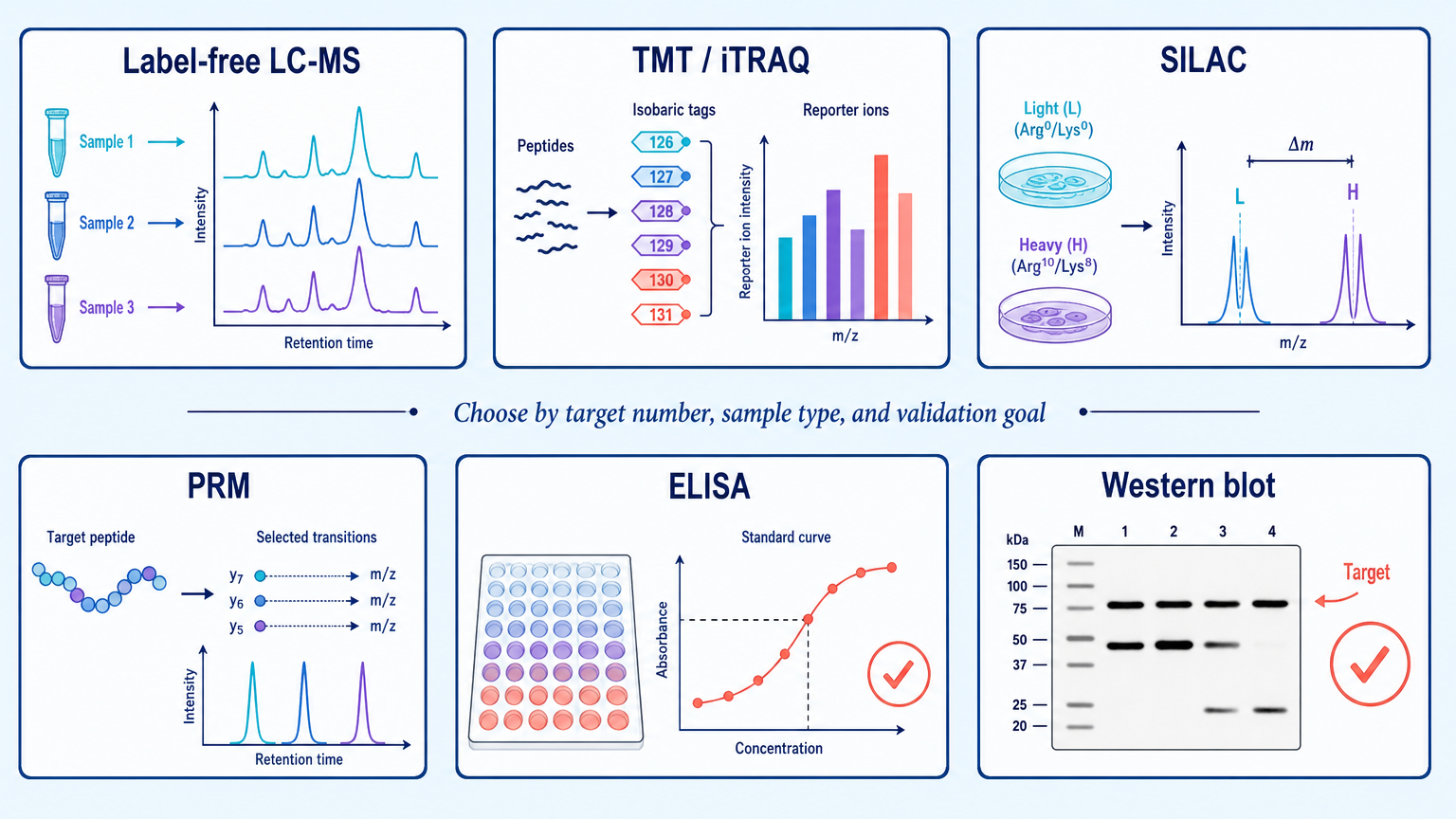
Label-free LC-MS is flexible for discovery studies. TMT and iTRAQ multiplex samples in a single experiment. SILAC is powerful in compatible cell culture systems. PRM is useful for targeted validation. ELISA and Western blot remain practical when high-quality antibodies are available and the target list is small.
Mechanism 5: Post-Translational Modification Analysis
Post-translational modifications change protein function, localization, stability, interaction behavior, and signaling output. Common targets include phosphorylation, glycosylation, acetylation, methylation, ubiquitination, oxidation, and many specialized modifications.
PTM analysis is harder than total protein analysis because modified peptides may be rare, labile, heterogeneous, or difficult to ionize. Enrichment is often needed. Phosphopeptide enrichment, glycopeptide enrichment, immunoprecipitation, specific digestion strategies, and targeted MS can all improve detection.
The interpretation should distinguish modified peptide abundance from total protein abundance. A phosphorylation site can increase even when the total protein amount does not change.
Structural and Interaction-Focused Protein Analysis
Some projects need information beyond identity and abundance. Intact protein analysis can measure molecular weight and proteoform patterns. Top-down or middle-down MS can preserve larger sequence context. Hydrogen-deuterium exchange MS, crosslinking MS, native MS, NMR, cryo-EM, and related methods can probe structure, conformational dynamics, or protein interactions.
These methods are not interchangeable. Structural analysis, interaction analysis, and abundance analysis answer different questions. The right method depends on whether the target is a purified protein, protein complex, cell lysate, clinical sample, or formulated biologic.
Main Applications
Protein analysis supports biomarker discovery, disease mechanism research, drug target identification, biopharmaceutical characterization, quality control, functional genomics, pathway analysis, and structural biology.
In discovery research, the goal is often to generate a reliable protein list and find biological patterns. In biopharmaceutical work, the goal is often more specific: confirm identity, purity, sequence coverage, glycosylation, degradation, aggregation, host cell proteins, or batch comparability.
Main Limitations
Protein analysis can fail for reasons that are easy to miss. Complex matrices suppress signal. Low-abundance proteins may fall below detection. Shared peptides complicate protein inference. Antibodies can cross-react. PTM enrichment can introduce bias. Large datasets can produce statistically significant results that are not biologically meaningful.
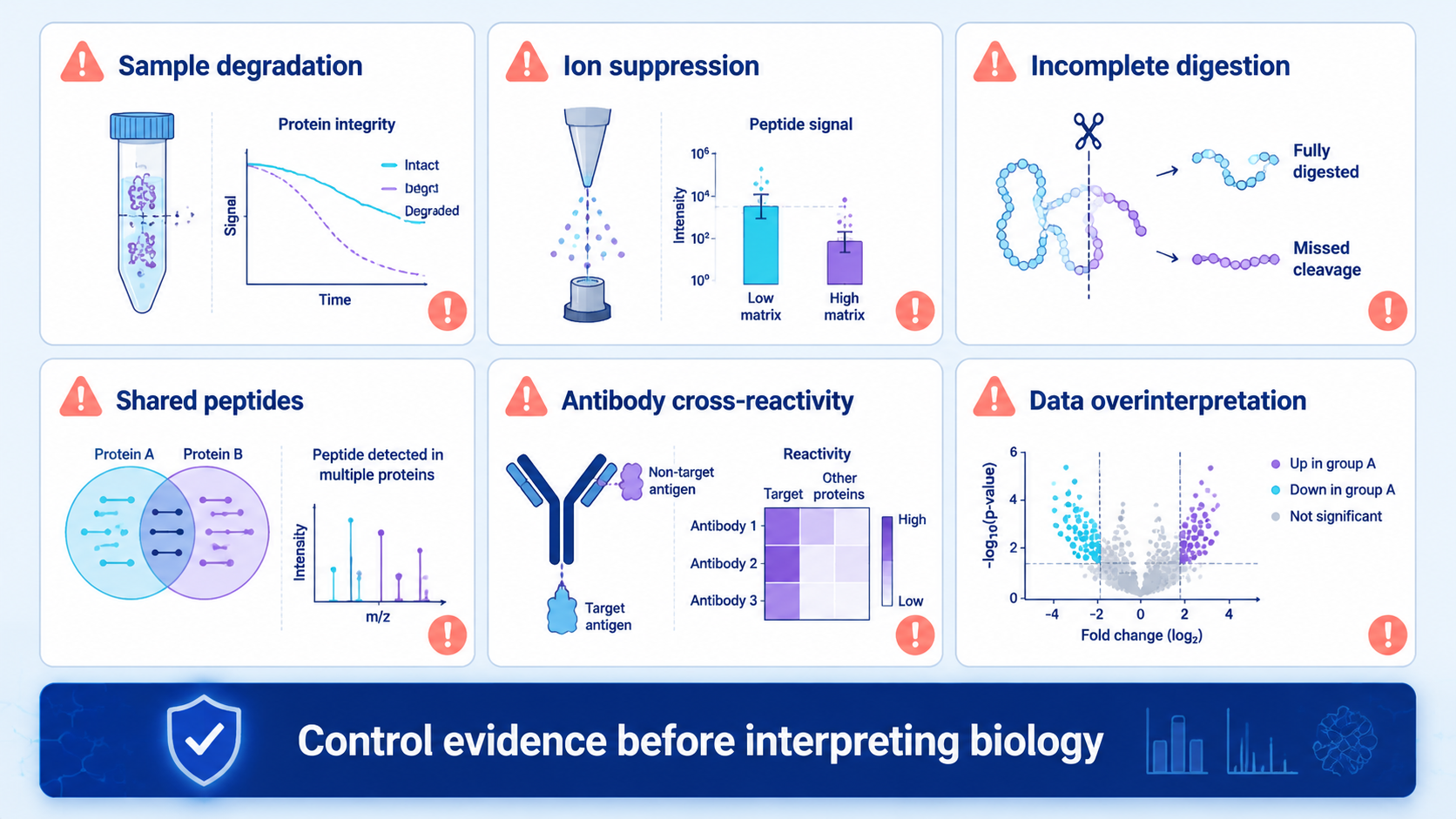
The safest workflow documents sample handling, digestion efficiency, instrument QC, search settings, peptide evidence, normalization, missing values, and validation criteria.
How to Choose a Protein Analysis Method?
| Research question | Better starting point | Why it fits | Main caution |
|---|---|---|---|
| What protein is in this band or sample? | LC-MS/MS protein identification | Sequence-based identification | Needs good peptide coverage |
| How much does protein abundance change? | Label-free, DIA, TMT/iTRAQ, or SILAC proteomics | Broad quantitative comparison | Batch design and normalization matter |
| Is one known protein present? | ELISA or Western blot | Practical targeted detection | Antibody specificity limits confidence |
| Which PTM sites are regulated? | Enriched PTM LC-MS/MS | Site-level modification evidence | Modified peptides may be low-abundance |
| What is the intact molecular weight? | Intact protein MS | Proteoform-level mass information | Complex mixtures can be difficult |
| Which candidates should be validated? | PRM or orthogonal assays | Targeted confirmation | Requires well-selected peptides or antibodies |
FAQ
1. What is the mechanism of protein analysis?
The mechanism of protein analysis is the workflow that converts proteins in a biological or biopharmaceutical sample into measurable signals. It usually includes extraction, separation, detection, identification, quantification, and computational interpretation.
2. Why is mass spectrometry widely used in protein analysis?
Mass spectrometry can identify proteins by peptide mass and fragmentation patterns, quantify abundance, and characterize modifications. It is especially useful when many proteins must be analyzed at once.
3. What is the difference between protein identification and protein quantification?
Protein identification asks which proteins are present. Protein quantification asks how much protein is present or how abundance changes across samples. The same LC-MS/MS experiment may support both, but the data analysis criteria are different.
4. When is antibody-based analysis better than LC-MS?
Antibody-based analysis is useful when the target protein is known, a specific antibody is available, and the project needs targeted detection across many samples. LC-MS is stronger when the target is unknown, many proteins are being screened, or PTM site information is needed.
5. Why is PTM analysis more difficult than total protein analysis?
PTM analysis is more difficult because modified peptides are often low-abundance, chemically diverse, and influenced by enrichment bias. Site localization and total protein abundance must also be interpreted separately.
Conclusion
Protein analysis works best when the method follows the biological question. Extraction and separation determine what reaches the detector. LC-MS/MS, antibody assays, structural methods, and PTM workflows each generate different kinds of evidence. A reliable study chooses the right mechanism, controls the sample preparation, and validates the results that matter most.
How to order?

