





Principles and Applications of De Novo Sequencing in Peptide Discovery and Characterization
- Use de novo sequencing when the sequence is unknown, poorly annotated, truncated, impurity-associated, or heavily modified.
- Start with LC-MS/MS evidence quality, especially precursor ion isolation, charge state behavior, and continuity of b ions and y ions.
- Expect outputs to range from a sequence tag to a near-complete candidate sequence, not automatic final confirmation.
- Plan orthogonal validation if the result will guide synthesis, impurity disposition, or a sequence-critical development decision.
- Unknown peptide identification from natural extracts, discovery fractions, or poorly annotated species
- Impurity characterization for synthetic peptides or biopharmaceutical products
- Investigation of a sequence variant that changes one or more residues
- Analysis of a post-translational modification (PTM)-rich analyte that fragments in a less predictable way
- Protein de novo sequencing projects where peptide-level evidence must be assembled because no complete reference sequence is available
- Select a precursor ion with adequate isolation quality.
- Generate a fragmentation spectrum using tandem mass spectrometry, often with collision-induced dissociation (CID) or higher-energy collisional dissociation (HCD).
- Inspect whether the spectrum contains a readable ion ladder.
- Match mass differences between adjacent fragment ions to amino acid residue masses.
- Build a sequence tag or candidate sequence while tracking gaps, competing explanations, and modification-related uncertainty.
- a full candidate sequence
- a partial sequence tag
- a localized or bounded PTM localization result
- confirmation of a suspected sequence variant
- a residue-supported explanation for an impurity-related mass feature
- Database search tests whether a known sequence explains the data.
- De novo sequencing tests which residue order the data support directly.
- Combined interpretation asks whether searchable candidates and spectral evidence converge.
- annotated fragmentation spectrum views
- one or more sequence tags
- ranked candidate sequence proposals
- notes on residue ambiguity
- comments on spectral annotation depth
- consistency checks against intact mass
De novo sequencing becomes relevant when an analyte cannot be identified with enough confidence by database search and the project needs direct residue-order evidence rather than the closest known match. In LC-MS/MS, the practical issue is not simply whether tandem mass spectrometry detects a peptide, but whether the fragmentation spectrum contains enough interpretable fragment ion information to support a sequence tag, a ranked sequence proposal, or an ambiguity-aware assignment.
Quick decision guide
What Problem De Novo Sequencing Solves
A standard database search works well when the true sequence is already present in the searchable reference space or differs only slightly from it. That assumption breaks down in several common project settings:
In those situations, a database search may return no match, weak confidence, or candidates that fit the precursor mass but not the observed fragmentation spectrum. At that point, the analysis shifts from reference matching to database-independent identification. The question is no longer which known sequence fits best, but which residue order the spectrum itself supports.
Four cause categories usually matter most when making that call.
Incomplete reference sequence space
If the analyte is novel, species-specific, engineered, degraded, or simply missing from the database, search scoring may fail even when the MS/MS data are otherwise usable.
Need for residue-level reconstruction
Some studies need more than a family-level assignment. A team may need to separate truncation from substitution, define a sequence variant, or determine whether a mass shift reflects a PTM or a sequence change.
Fragmentation complexity
Mixed precursor isolation, neutral losses, internal ions, and partial ion ladders can all lower database-search confidence. Even so, the same spectra may still support a useful sequence tag when interpreted with de novo logic.
Intrinsic MS/MS ambiguity
Some limits come from the evidence itself rather than from the software. Leucine/Isoleucine ambiguity is a classic example because those residues are isobaric in routine MS/MS interpretation. PTMs, cyclization, disulfide constraints, and incomplete fragmentation can also leave residue ambiguity in otherwise informative spectra.
Principle of De Novo Sequencing in LC-MS/MS
In peptide de novo sequencing, amino acid order is inferred directly from tandem mass spectrometry rather than assigned only through comparison with a sequence database. The core evidence comes from the mass differences between consecutive fragment ion signals, especially b ions and y ions, which reflect peptide backbone cleavage.
A simplified interpretation flow looks like this:
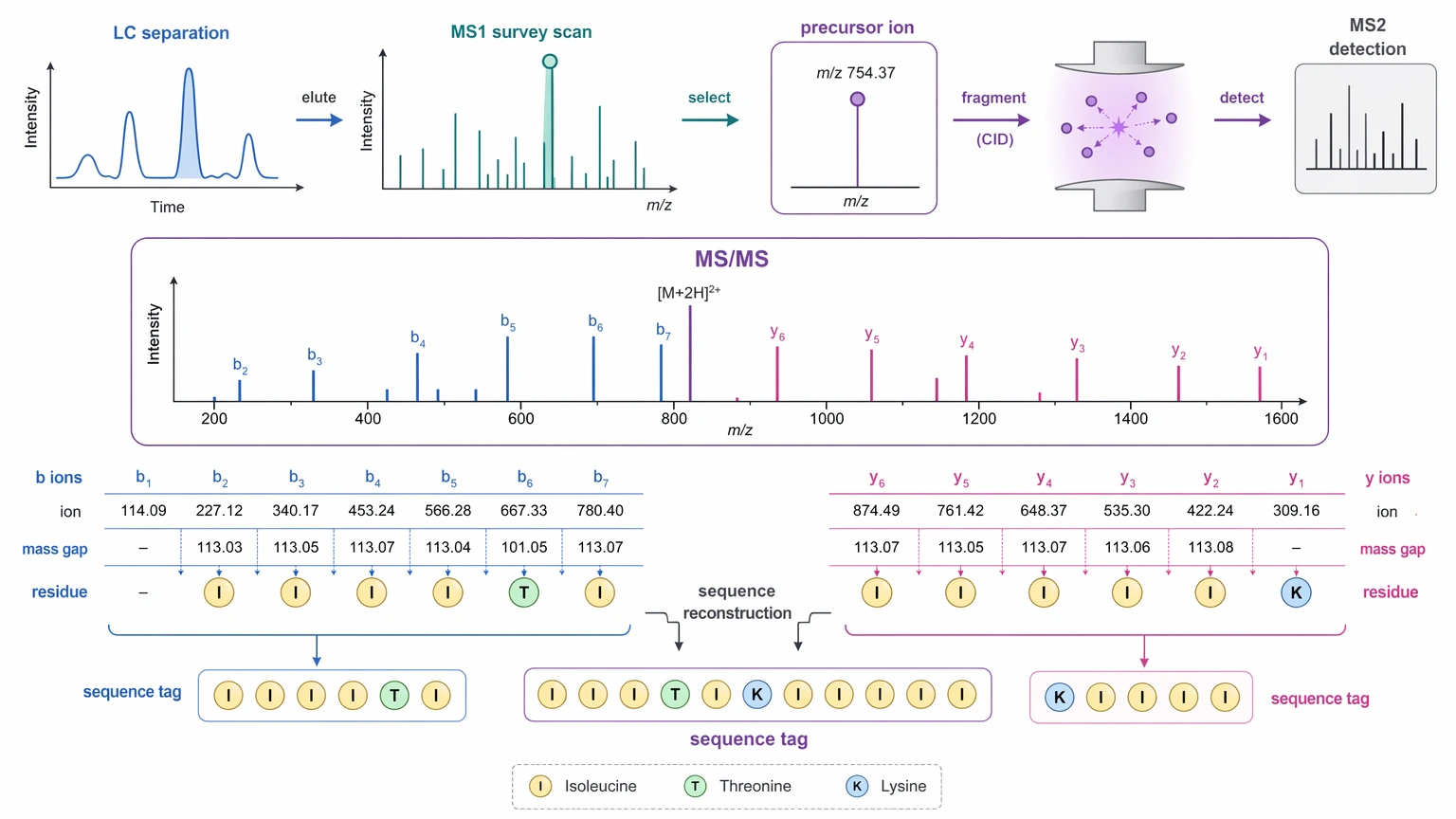
This is useful because a sequence can still be proposed even when the correct entry is missing from a database. Its main limitation is just as direct: the proposal is only as strong as the fragmentation evidence behind it.
An explicit limitation matters here: MS/MS-based de novo sequencing may not resolve every residue or PTM position with high confidence, especially when ion ladders are incomplete, multiple PTMs are present, or database-independent proposals must be interpreted from mixed or low-abundance spectra.
How to Decide Whether the Method Fits Your Project
For a method-selection article, the most practical starting point is to define the output you actually need, then judge whether the sample and spectra can support it.
Step 1: Define the deliverable you actually need
Not every project requires the same level of sequence certainty. The target deliverable may be:
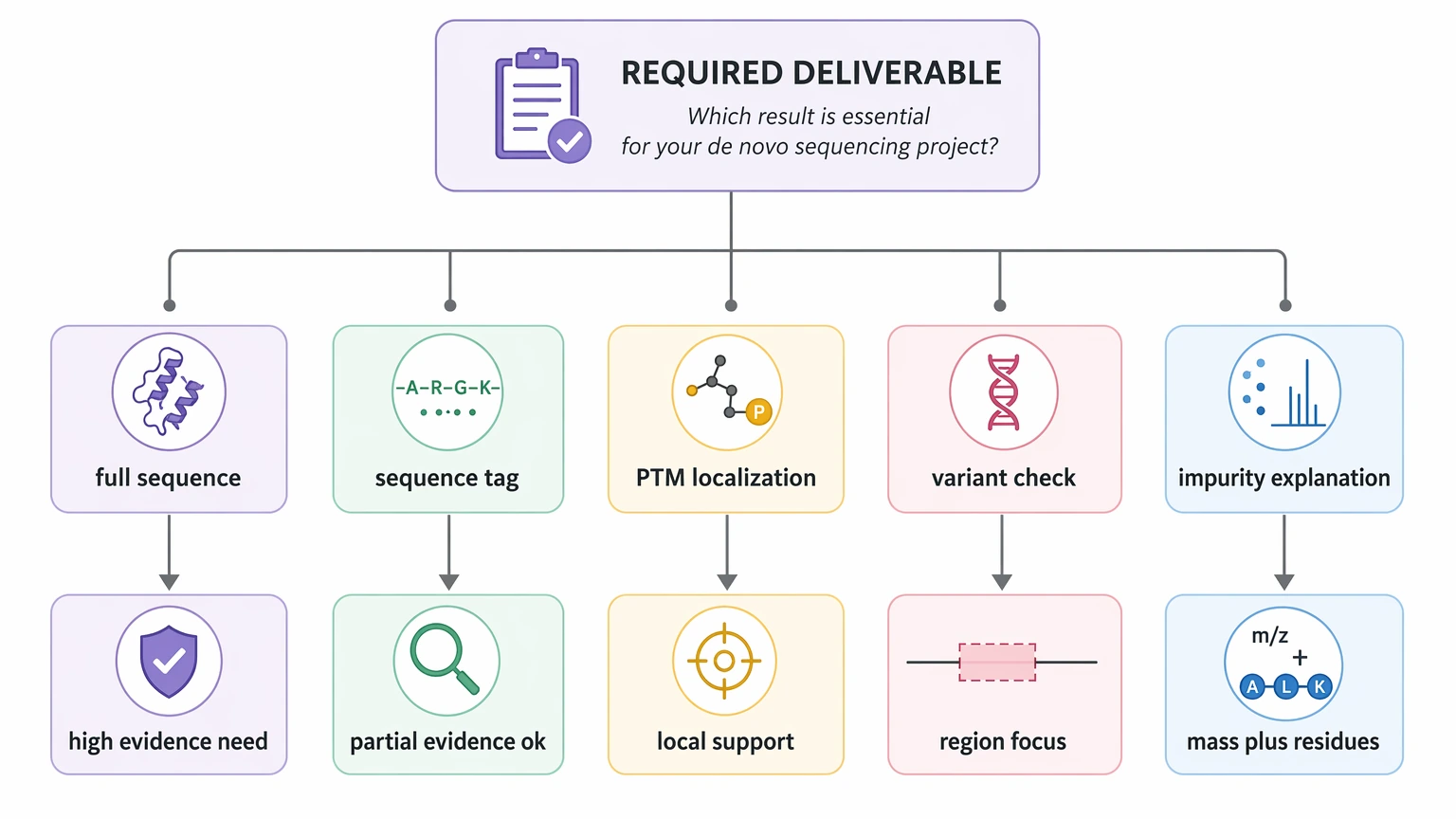
That distinction changes whether de novo sequencing is the right fit. For example, impurity characterization may only need evidence for truncation plus intact mass agreement, while a peptide discovery project may need a longer residue series to support synthesis or follow-up testing.
Step 2: Check whether the spectra are informative enough
The table below helps separate promising cases from weak-input cases.
| Scenario | Recommended workflow | Key limitation | Validation need |
|---|---|---|---|
| Unknown peptide with no credible database match | De novo sequencing from LC-MS/MS fragmentation spectrum | Incomplete ion series may prevent full assignment | Intact mass and targeted follow-up |
| Peptide impurity with unexpected mass shift | Database search plus de novo sequencing | Low abundance or co-isolation can blur spectral annotation | Replicate spectra and intact mass |
| Suspected sequence variant in a known product family | Constrained search plus local de novo interpretation | Single-residue ambiguity may remain | Targeted LC-MS/MS confirmation |
| PTM-rich analyte with weak search scores | Modification-aware de novo sequencing | PTM localization may stay partial | Complementary fragmentation or orthogonal validation |
| Poorly annotated protein | Protein de novo sequencing from peptide-level evidence | Coverage gaps across peptides | Multi-peptide consistency review |
The most informative spectra usually show clean precursor isolation, workable charge states, and enough continuity in b ions or y ions to support residue-to-residue transitions. Warning signs include broad co-elution, strong matrix interference, and dominant mixed spectra.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
A short project-fit discussion can save time here. If your team needs to decide whether existing spectra, sample amount, and characterization goals support a de novo workflow, you can submit your requirements to MtoZ Biolabs to evaluate your project before committing to a larger sequencing and validation plan.
Step 3: Decide whether de novo sequencing should complement or lead the workflow
In many studies, de novo sequencing does not replace database search. It works alongside it.
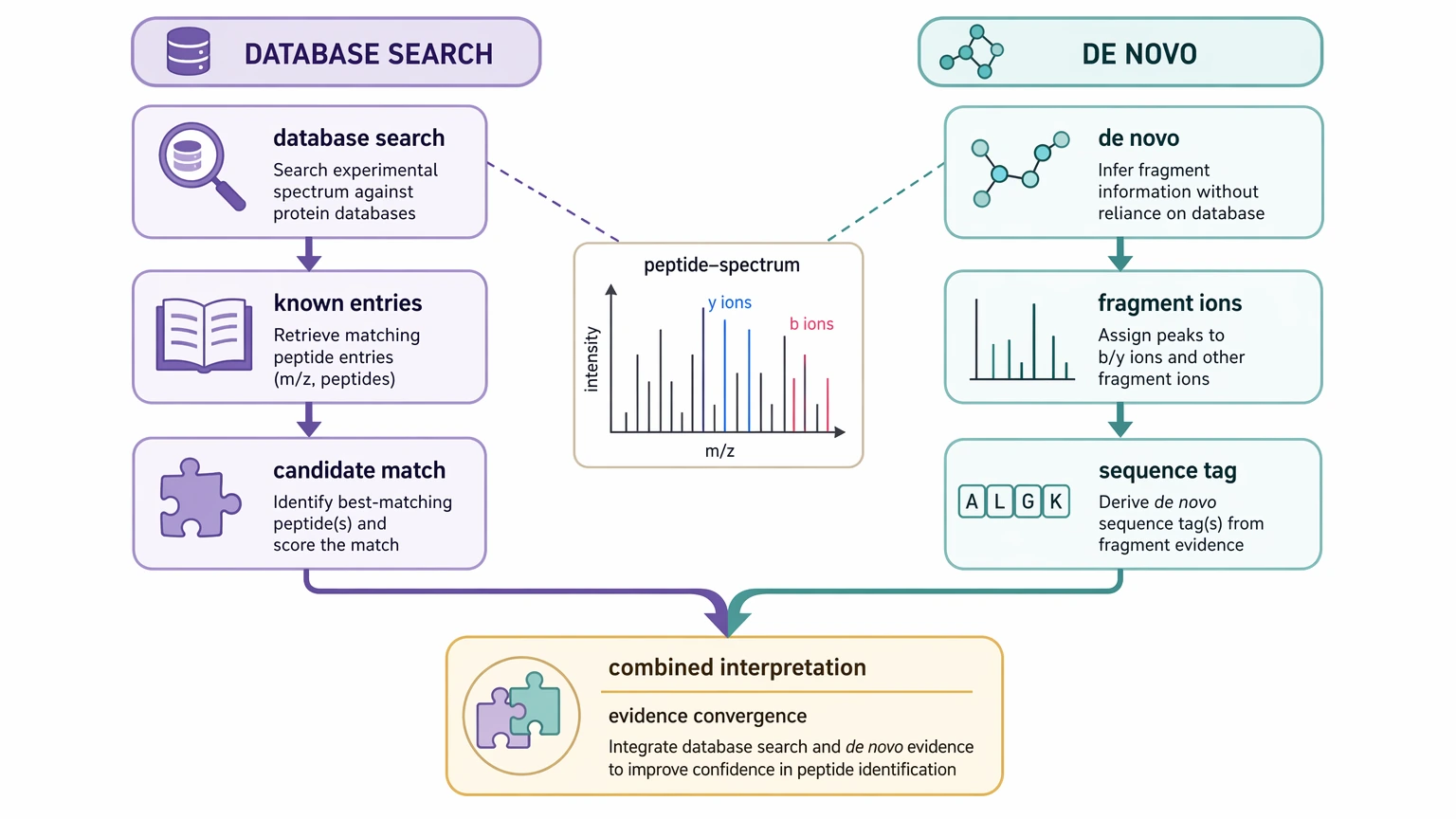
That combined approach is often the most efficient option in peptide characterization, especially when the project starts with some prior knowledge but not enough for confident reference matching.
Where De Novo Sequencing Is Applied
The method is most useful in projects where direct sequence evidence changes the next decision.
Peptide discovery
Novel bioactive fractions, secreted peptides, and natural product-associated peptide mixtures may not map well to available databases. A sequence tag can narrow the candidate space and guide synthesis or follow-up isolation.
Impurity characterization
Unexpected peaks in synthetic or formulated peptide products often require a residue-level explanation. De novo sequencing can separate truncation, substitution, modification, or mixed-species signals when precursor mass alone is not enough.
Sequence variant review
When a known product family contains a suspected sequence variant, local de novo interpretation can clarify whether the altered region is actually supported by the MS/MS evidence.
PTM-rich peptide characterization
A post-translational modification (PTM) may be the reason sequencing is needed and also the reason it becomes harder. Labile PTMs can change fragmentation behavior, and PTM localization may remain uncertain unless multiple fragment ions support the same site assignment.
Protein de novo sequencing
In practice, protein de novo sequencing usually means assembling evidence from multiple peptides rather than reading an intact protein sequence in a single step. That makes coverage consistency and cross-peptide interpretation central to confidence.
Expected Results and Validation Methods
A realistic de novo sequencing result is an evidence package, not just a software-generated sequence string. It helps to separate immediate outputs from follow-up confirmation.
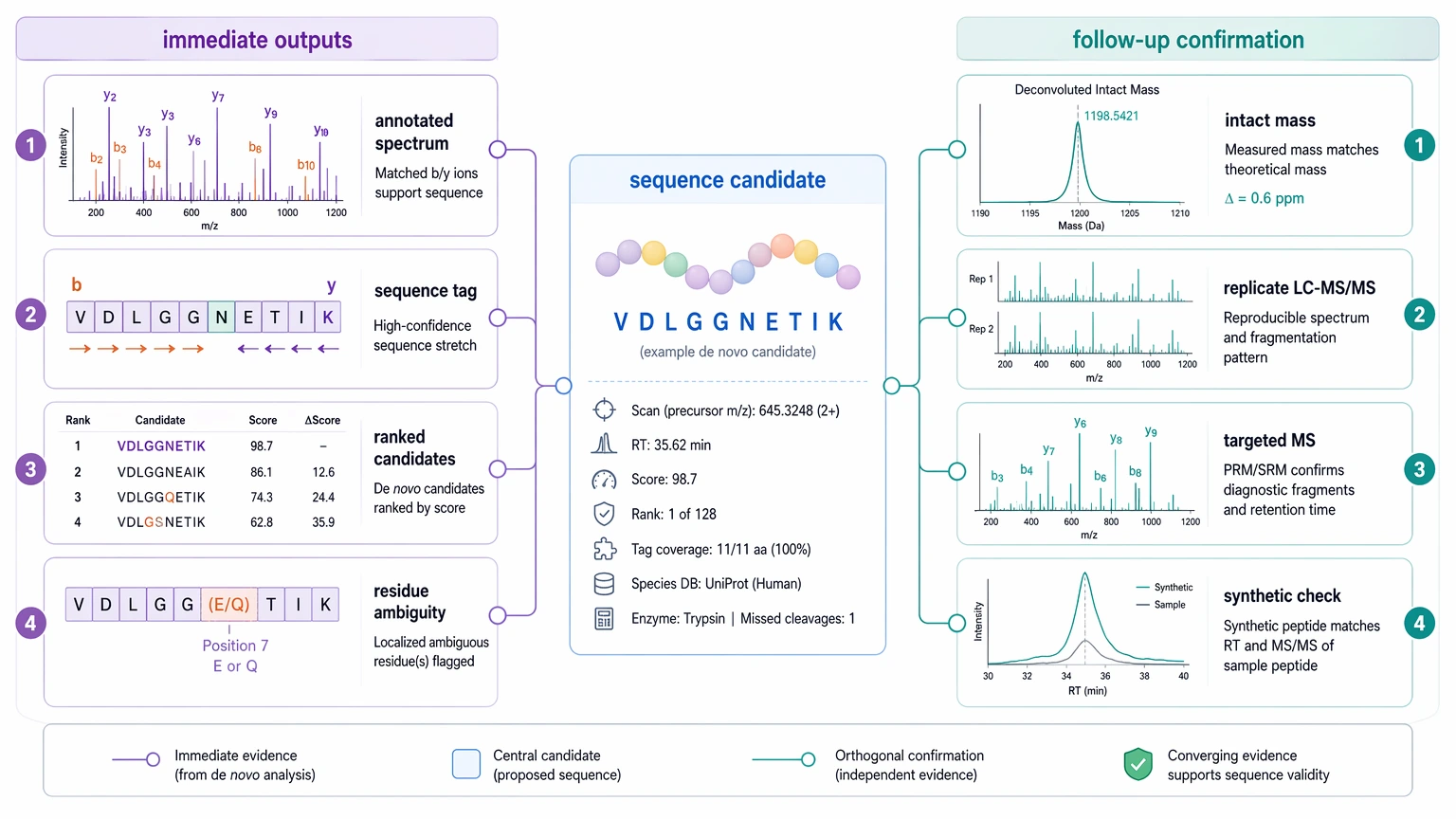
Immediate deliverables
Immediate de novo deliverables often include:
Follow-up confirmation
Follow-up confirmation adds the next layer of evidence when the sequence assignment will affect a development, impurity, or decision-making milestone.
| Evidence type | Immediate deliverable or follow-up confirmation | What it supports | Main boundary |
|---|---|---|---|
| Annotated b/y ion ladder | Immediate deliverable | Residue connectivity across a sequence region | Gaps reduce local confidence |
| Sequence tag | Immediate deliverable | Partial database-independent identification | May not define full residue order |
| Intact mass agreement | Follow-up confirmation | Composition-level consistency | Does not prove sequence order |
| Replicate LC-MS/MS spectra | Follow-up confirmation | Reproducibility of sequence features | Cannot remove intrinsic isobaric ambiguity |
| Complementary fragmentation or targeted MS | Follow-up confirmation | Stronger support for variant or PTM localization | Still depends on interpretable spectra |
| Synthetic peptide or Edman comparison | Follow-up confirmation | Higher-confidence verification of a proposed candidate | Requires a narrowed candidate set |
Use this distinction carefully: a ranked sequence proposal can be scientifically useful before it is fully confirmed. The right next step depends on whether the project needs partial identification, impurity explanation, or a tighter sequence-level conclusion.
Key Cautions and Practical Limits
Before choosing this route, it helps to keep the main operational limits in view.
Sample quality and amount can limit interpretability
Low abundance, poor enrichment, or strong matrix background can limit the output to short sequence tags. Even when the method fits the question, the available sample may not support near-complete assignment.
Controls and repeat measurements still matter
Replicate acquisition helps show whether a proposed sequence feature is reproducible or driven by a weak spectrum. In impurity work, reference material or process context may add as much value as the MS/MS data itself.
Batch effects, carryover, and contamination can mimic complexity
Co-isolation, carryover, and mixed peptide populations can create misleading fragmentation patterns. When precursor purity is uncertain, interpretation should remain conservative.
Interpretation has clear boundaries
De novo sequencing does not automatically solve Leucine/Isoleucine ambiguity, every PTM problem, or every protein-level gap. A single spectrum may support a useful hypothesis while still leaving unresolved residue positions or alternative explanations.
Another method may be the better next step
If the main question is total molecular composition, intact mass may be the faster first step. If the N-terminus is accessible and the sequence question is narrow, Edman comparison may be more direct. If the main obstacle is poor spectral quality rather than an unknown sequence burden, better LC-MS/MS acquisition may help more than deeper interpretation alone.
If your team is weighing those tradeoffs, contact MtoZ Biolabs to discuss the study with sample context, intact mass data, and representative spectra so the next method can be matched to the actual decision point rather than to a generic sequencing request.
Conclusion
De novo sequencing is most informative when a project needs sequence-level evidence that database search cannot provide with enough confidence. In peptide discovery, impurity characterization, sequence variant review, and PTM-rich peptide characterization, its value comes from direct interpretation of fragment ion patterns rather than reliance on a complete reference library. For unknown or poorly annotated analytes, the best-fit projects are those with interpretable LC-MS/MS data, a clear need for residue-order evidence, and a validation plan that separates immediate sequence proposals from later confirmation. When those conditions are in place, de novo sequencing can offer a practical route to peptide characterization. When they are not, the better next step may be improved separation, cleaner spectra, intact mass support, or an alternative confirmation method.
FAQ
Can de novo sequencing still help if database search returns a weak match instead of no match?
Yes. A weak match can still be useful context, but de novo sequencing can test whether the observed residue transitions support that candidate or instead point to a truncation, modification, or different sequence region.
What peptide length range is hardest for de novo interpretation?
Very short peptides can lack enough informative fragment spacing, while longer peptides often show more gaps, mixed fragmentation behavior, or overlapping interpretations. The difficult range depends on spectrum quality and modification burden, not just peptide length.
Does a higher-resolution instrument automatically remove residue ambiguity?
No. Higher resolution improves mass accuracy and can sharpen spectral annotation, but it does not by itself resolve every isobaric case, especially Leucine/Isoleucine ambiguity or poorly localized PTMs.
When is partial sequence information enough to move a project forward?
A partial sequence tag may be enough when the goal is to narrow candidate space, explain a truncation, group an analyte into a peptide family, or choose a targeted confirmation strategy rather than claim full sequence confirmation.
Should I sequence the intact protein or digest it first for protein de novo sequencing?
For many proteins, peptide-level evidence from digests is more practical because it tends to produce more interpretable fragmentation spectra. Intact protein approaches may help in some contexts, but they do not replace peptide-level reconstruction in most de novo projects.
What information is most useful before starting a feasibility discussion?
Prepare the analyte source, sample amount, enrichment or purity status, expected molecular mass range, any intact mass result, and a few representative MS/MS spectra. That usually makes the first workflow decision much more precise.
How to order?

