





Planning Protein De Novo Sequencing for Blocked N-Termini, Sequence Variants, or Low-Reference Samples
- confirming whether a purified protein differs from the expected construct
- characterizing internal sequence when the N-terminus is blocked
- finding a candidate sequence variant in an engineered or processed protein
- generating de novo peptide sequencing evidence for a low-reference sample
- separating PTM-driven mass shifts from true amino acid substitution events
- Is the protein a dominant species or part of a mixed preparation?
- Is an intact mass measurement already available?
- How much material can be reserved for repeat LC-MS/MS runs or orthogonal validation?
- Is the buffer compatible with protease digestion and tandem mass spectrometry?
- Is heterogeneity expected from glycosylation, oxidation, cleavage, or formulation components?
- broad internal sequence tag recovery
- localization of a specific amino acid substitution
- comparison of PTM and sequence-variant explanations
- better confidence around an uncertain terminal region
- de novo peptide sequencing results with annotated sequence tags
- internal peptide coverage maps
- overlapping peptides that support continuous regions
- confidence annotation for uncertain residues or localized variants
- PTM-aware notes where modification and substitution interpretations compete
- explicit flags for leucine/isoleucine ambiguity or unresolved terminal regions
- intact mass consistency checks against reconstructed regions
- peptide mapping against newly inferred candidate sequences
- terminal analysis when the blocked N-terminus is central to the question
- targeted re-analysis of a proposed sequence variant from an independent digest
- protein source, construct background, and expected molecular weight
- whether a blocked N-terminus or known N-terminal modification is suspected
- what reference sequence coverage exists and how close the homologous sequence is
- whether the concern is a sequence variant, truncation, PTM, or low-reference identification
- available sample amount and purity context
- any existing intact mass or peptide mapping data
- the exact decision the final report must support
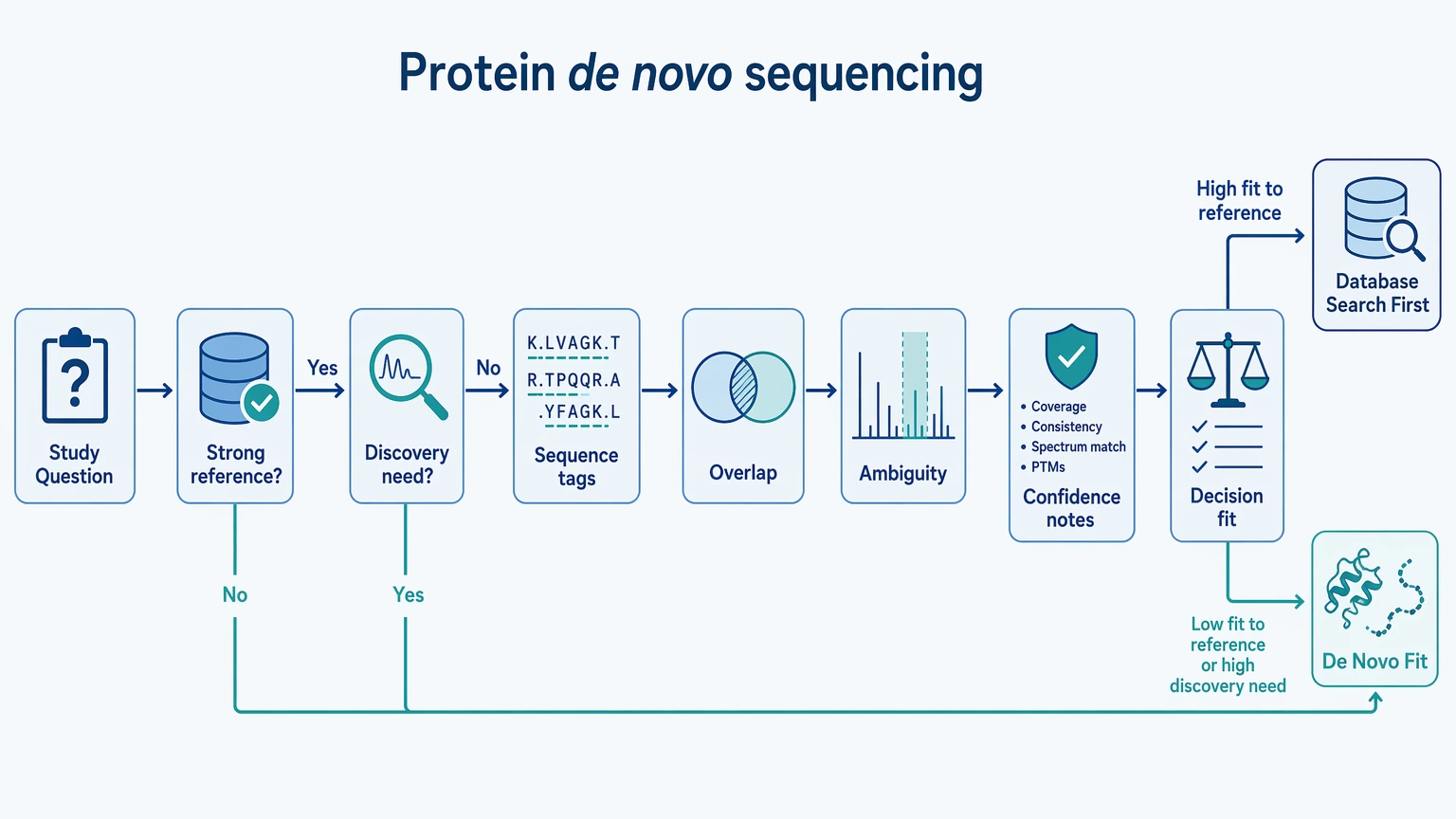
Protein de novo sequencing is most useful when the real task is sequence reconstruction rather than routine protein identification. That tends to be the case when a blocked N-terminus limits direct terminal characterization, when a sequence variant or amino acid substitution may be present, or when reference coverage is too weak for database searching alone to support a confident answer. The planning work starts before LC-MS/MS acquisition. You need to decide what has to be resolved, how much sample you can afford to spend on repeat analysis, and how much ambiguity the final report can tolerate.
Quick Decision Guide
Use protein de novo sequencing as the primary workflow when the expected sequence may be missing, divergent, or masked by N-terminal modification.
Use database search and peptide mapping first when the reference is strong and the main goal is confirmation, not discovery.
Plan orthogonal validation early if the project depends on terminal assignment, variant localization, or residue-level certainty in a low-reference region.
A practical way to frame the choice is to ask whether your project can work with a report built from sequence tags, overlapping peptides, ambiguity zones, and confidence annotations instead of one uninterrupted, fully certain sequence string. If that level of evidence is enough to answer the study question, protein de novo sequencing is often the better fit.
Where Standard Identification Breaks Down
These studies usually run into a few specific technical problems, not a broad failure of tandem mass spectrometry.
First, database search loses power when the true sequence is not represented well enough. A distant homolog can still give useful context, but it may not support confident calls for substitutions, truncations, or unusual terminal regions. In low-reference samples, the top database hit may point you in the right direction while still being wrong at the residue level.
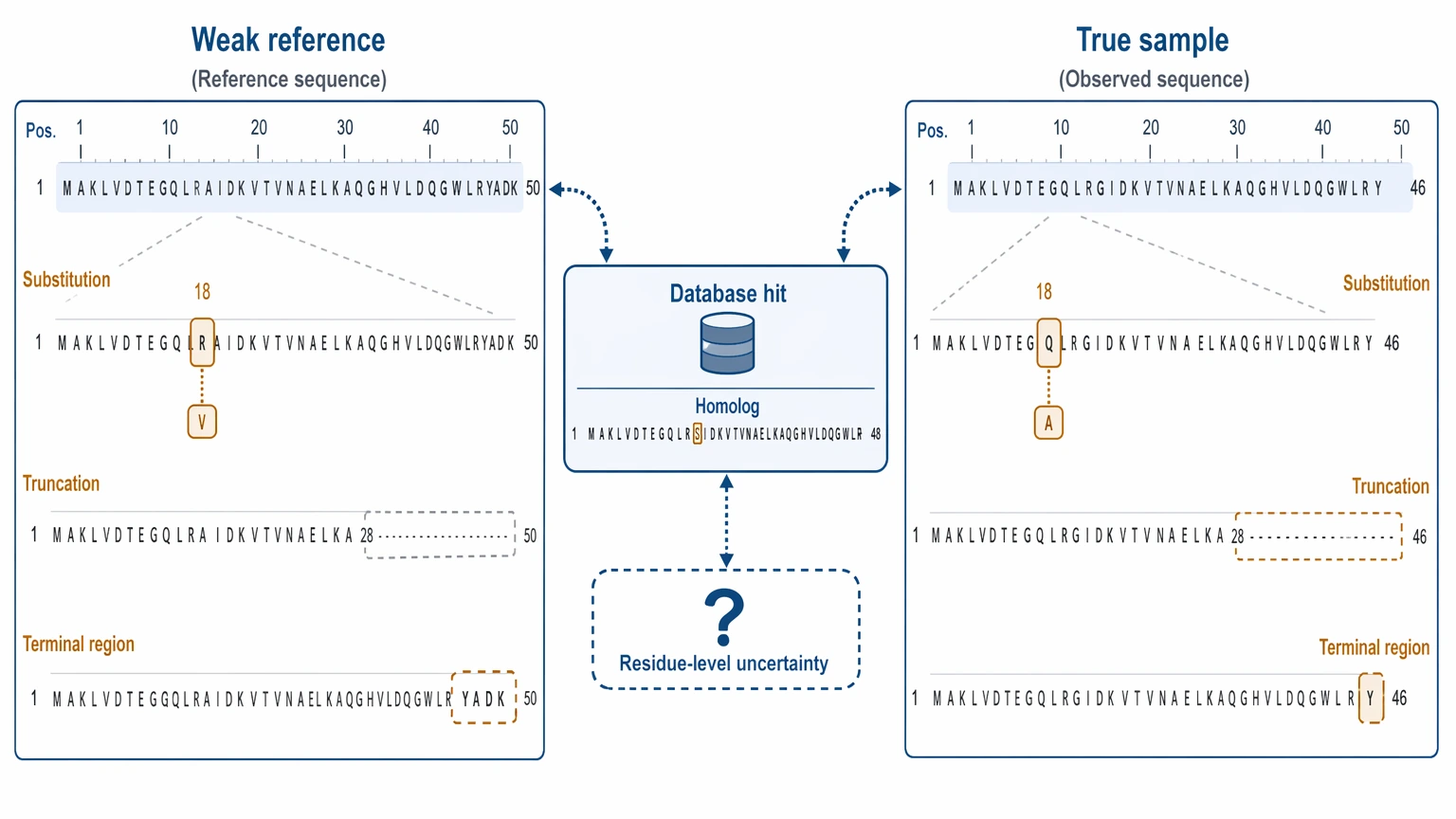
Second, a blocked N-terminus changes the normal sequencing logic. If the protein start carries pyroglutamate, acetylation, or another N-terminal modification, direct N-terminal readout may be limited or only inferred indirectly. In that situation, internal peptide coverage and peptide overlap matter more than the missing terminus itself.
Third, single-digest evidence is often too fragmented for sequence reconstruction. Routine identification can work with many isolated peptide IDs. Protein de novo sequencing usually needs overlapping peptides that connect short local tags into longer, interpretable regions.
Fourth, PTMs can mimic sequence change. Oxidation, glycation, truncation, or processing heterogeneity may create mass shifts that look like sequence variants. Before calling an amino acid substitution, the data need PTM-aware interpretation.
Finally, some uncertainty is built into MS-based sequencing. Leucine/isoleucine ambiguity often remains unresolved by standard LC-MS/MS alone, and terminal confidence may stay incomplete even when internal evidence is strong. That limit should be written into the project plan up front, not discovered at the end.
A Planning Framework for Difficult Samples
This article follows a planning structure because the main goal is to set the workflow before the sample is used up.
Step 1: Define the exact question the report must answer
Start with the decision, not the platform. Common examples include:
If the project only needs to confirm a known protein against a strong reference, database search plus peptide mapping may still be the better first step. If the project requires sequence discovery under weak reference support, protein de novo sequencing makes more sense.
The table below helps match the problem to the right starting workflow.
| Scenario | Recommended workflow | Key limitation | Validation need |
|---|---|---|---|
| Known protein, strong reference support | Database search + peptide mapping | May miss undocumented substitutions | Intact mass if discrepancy remains |
| Blocked N-terminus, internal sequence needed | Protein de novo sequencing with overlap-focused digestion | Sequence start may remain indirect | Terminal follow-up or intact mass |
| Suspected sequence variant in engineered protein | De novo-supported variant review | Single-spectrum evidence is weak | Orthogonal validation of the site |
| Low-reference or poorly annotated sample | Protein de novo sequencing + homolog alignment | Full-length continuity may stay incomplete | Confidence annotation + follow-up confirmation |
Use this comparison to decide whether you need identification, reconstruction, or a staged combination of both. If your team is making a go/no-go call on a scarce sample, submit your requirements early so the study can be scoped around the sequence question rather than a default workflow.
Step 2: Check whether the sample is ready for interpretation
For difficult proteins, concentration alone does not tell you much. More useful questions include:
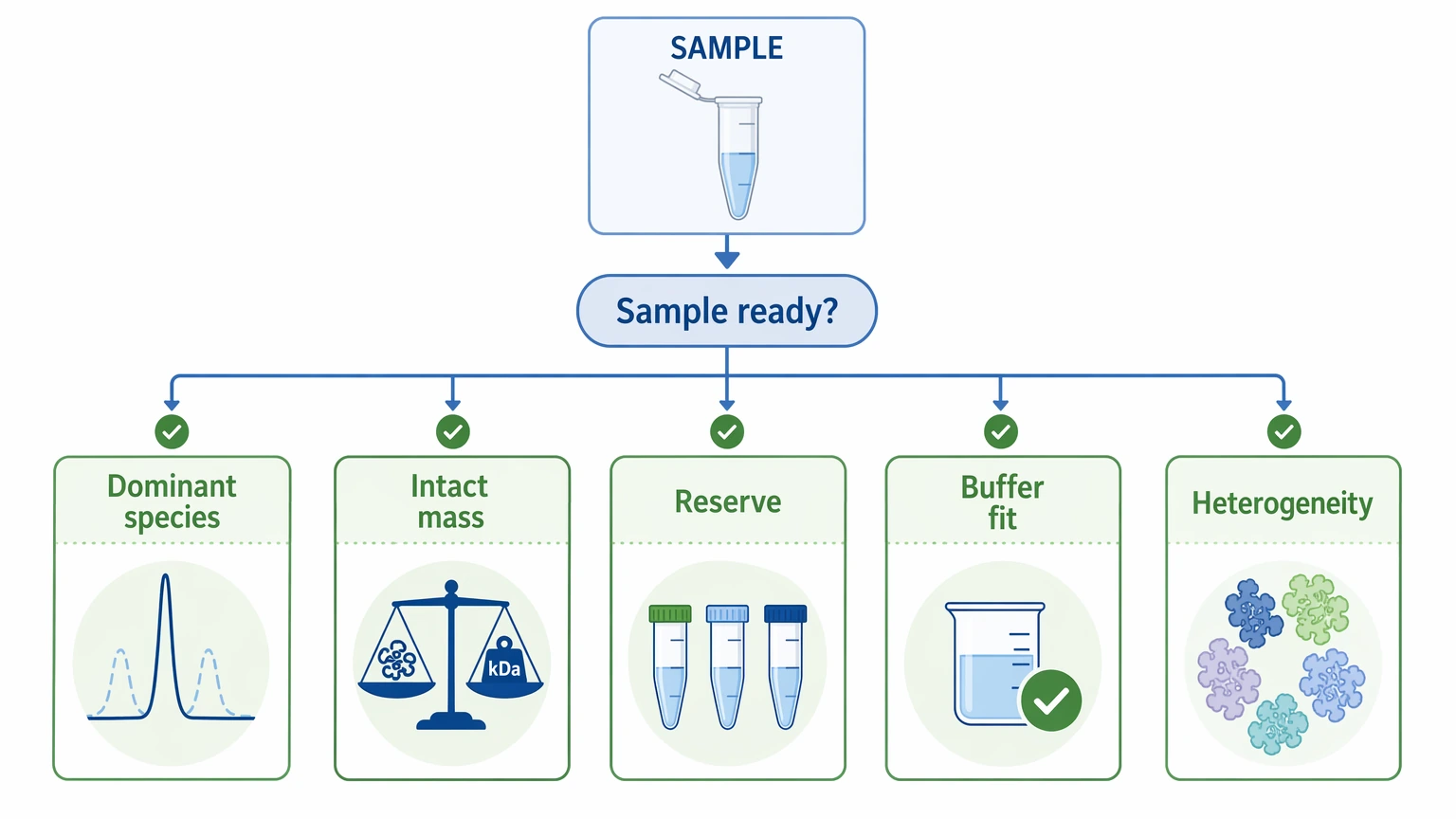
A partially purified sample can still be analyzed, but interpretation gets harder fast when multiple proteoforms or contaminants are present. In those settings, internal peptide coverage may reflect more than one species, which makes sequence assembly less secure.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
Step 3: Design protease digestion for overlap
For protein de novo sequencing, digestion strategy should support overlapping peptides, not just peptide count. Multi-enzyme digestion often improves continuity because it creates different cleavage patterns across the same region.
| Sample type | Best fit | Constraint | Next step |
|---|---|---|---|
| Purified protein with adequate material | Multi-enzyme digestion | Consumes more sample | Keep reserve for repeats |
| Limited sample, narrow question | Targeted digest emphasis | Less global coverage | Focus on the decision-critical region |
| Blocked N-terminus | Internal overlap from complementary enzymes | Terminus may stay unresolved | Pair with orthogonal terminal analysis if needed |
| PTM-rich protein | Modification-aware digest review | More complex spectra | Compare PTM and substitution models |
The aim is not maximum complexity. The aim is to generate sequence tags that connect across the region that matters most for the project.
Step 4: Align fragmentation strategy with the evidence you need
A fragmentation spectrum that is enough for routine identification is not always enough for de novo peptide sequencing. For sequence reconstruction, fragment-ion support has to extend the sequence tag, support residue order, and help localize a sequence variant or PTM.
Before starting, define whether the priority is:
This is also the point where expectations should be set clearly. LC-MS/MS can provide strong local evidence without proving every residue in a full-length sequence. Database-search limits, PTM burden, and incomplete fragmentation can all reduce confidence in specific regions.
Expected Results and Validation Strategy
A realistic protein de novo sequencing report often includes outputs at different levels of certainty rather than one clean final answer with no caveats.
Immediate deliverables
The first deliverables should usually include:
Follow-up confirmation
Keep those immediate outputs separate from later confirmation work. Follow-up confirmation may include:
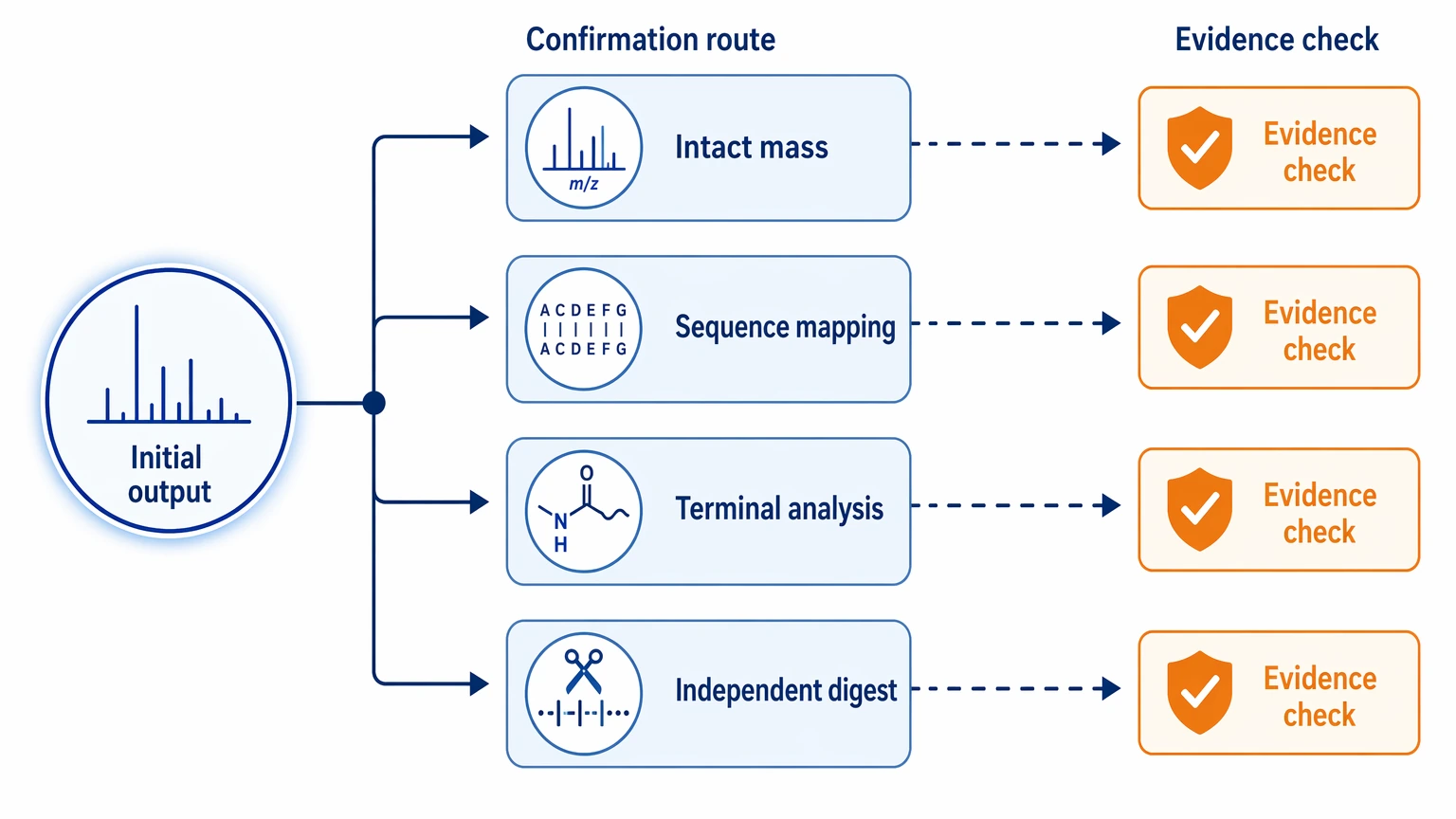
That distinction matters. A candidate substitution supported by one fragmentation spectrum is not the same as a confirmed site assignment backed by orthogonal validation. When a project depends on a terminal call or a low-reference region, plan the confirmation work in parallel instead of leaving it for later.
Key Cautions and Practical Limits
Protein de novo sequencing can be very informative in the right setting, but the boundaries need to be defined before sample submission.
Sample quality or amount limits: Multi-enzyme digestion, repeat LC-MS/MS runs, and orthogonal validation all consume material. Keep a reserve aliquot whenever possible, especially if terminal follow-up may be needed.
Controls and repeat expectations: If a sequence variant could affect development or characterization decisions, look for repeat support from an independent digest or separate acquisition, not only one favorable spectrum.
Batch and contamination risk: Keratin, buffer carryover, oxidation artifacts, and mixed protein populations can complicate sequence-tag interpretation. The risk is higher in low-input or partially purified samples.
Interpretation boundaries: Protein de novo sequencing may give high-confidence local regions while leaving uncertainty at specific residues or termini. Leucine/isoleucine ambiguity, PTM interference, and limited reference coverage are normal reporting limits, not signs that the analysis failed.
When another method is the better next step: If the decision depends on direct terminal assignment, a single exact substitution call, or attribution within a complex mixture, a staged workflow may be more efficient than de novo sequencing alone. In that case, you can contact MtoZ Biolabs to evaluate your project around sample readiness, workflow design, and the most useful validation sequence before committing the full sample set.
How to Prepare a Pre-Consultation Brief
A short planning brief can prevent wasted runs and unclear expectations. Include:
That level of detail helps align digestion design, LC-MS/MS strategy, and the validation plan with the actual scientific question.
Conclusion
Protein de novo sequencing makes the most sense when standard database search cannot answer the real question because terminal access is blocked, sequence drift is plausible, or reference support is too weak to trust a routine match. The strongest studies are planned around the evidence they actually need: overlapping peptides for continuity, confidence annotation for ambiguity, and orthogonal validation for claims that affect downstream decisions. For purified proteins, engineered variants, and low-reference samples where internal sequence evidence matters more than a database label, a structured pre-submission plan usually saves both material and interpretation time. If you are preparing a difficult characterization study, contact MtoZ Biolabs to submit your requirements and evaluate your project in the context of sample condition, expected deliverables, and the follow-up confirmation your team will actually need.
FAQ
Can de novo peptide sequencing still help if I only care about one short region?
Yes. If the decision depends on one uncertain region, the workflow can be designed around that segment rather than around full-length reconstruction. In that case, digestion and fragmentation choices should be judged by whether they improve overlap and fragment support in the region of interest.
Does a blocked N-terminus always mean the sequence start cannot be assigned?
No. It means the start may not be read directly. Sometimes the N-terminus can still be inferred through internal peptide evidence, intact mass context, or targeted terminal follow-up, but the confidence level should be reported clearly.
How do I tell whether a mass shift is a PTM or a true sequence variant?
Look at the fragmentation spectrum, localization support, and whether a PTM-aware explanation fits the data at least as well as a substitution model. A credible variant call usually has support across multiple ions or related peptides, not just a single mass difference.
Is a distant homolog useful in a low-reference project?
Often yes, but mainly as a scaffold for interpretation. A homologous sequence can help organize de novo sequence tags, yet it should not be treated as proof that the sample matches that homolog at every residue.
When should I reserve sample for orthogonal validation instead of spending everything on LC-MS/MS depth?
Reserve material when the project outcome depends on a terminal assignment, a localized substitution, or a region where the final sequence confidence must be stronger than de novo evidence alone can provide.
What does a strong de novo report look like if it does not give a complete sequence?
A strong report can still support decisions if it defines high-confidence sequence regions, shows overlapping peptides, marks ambiguity zones clearly, and states what confirmation step would strengthen the remaining uncertain calls.
How to order?

