Peptide and Protein De Novo Sequencing by Mass Spectrometry: How Sample Type Changes the Workflow and Readout
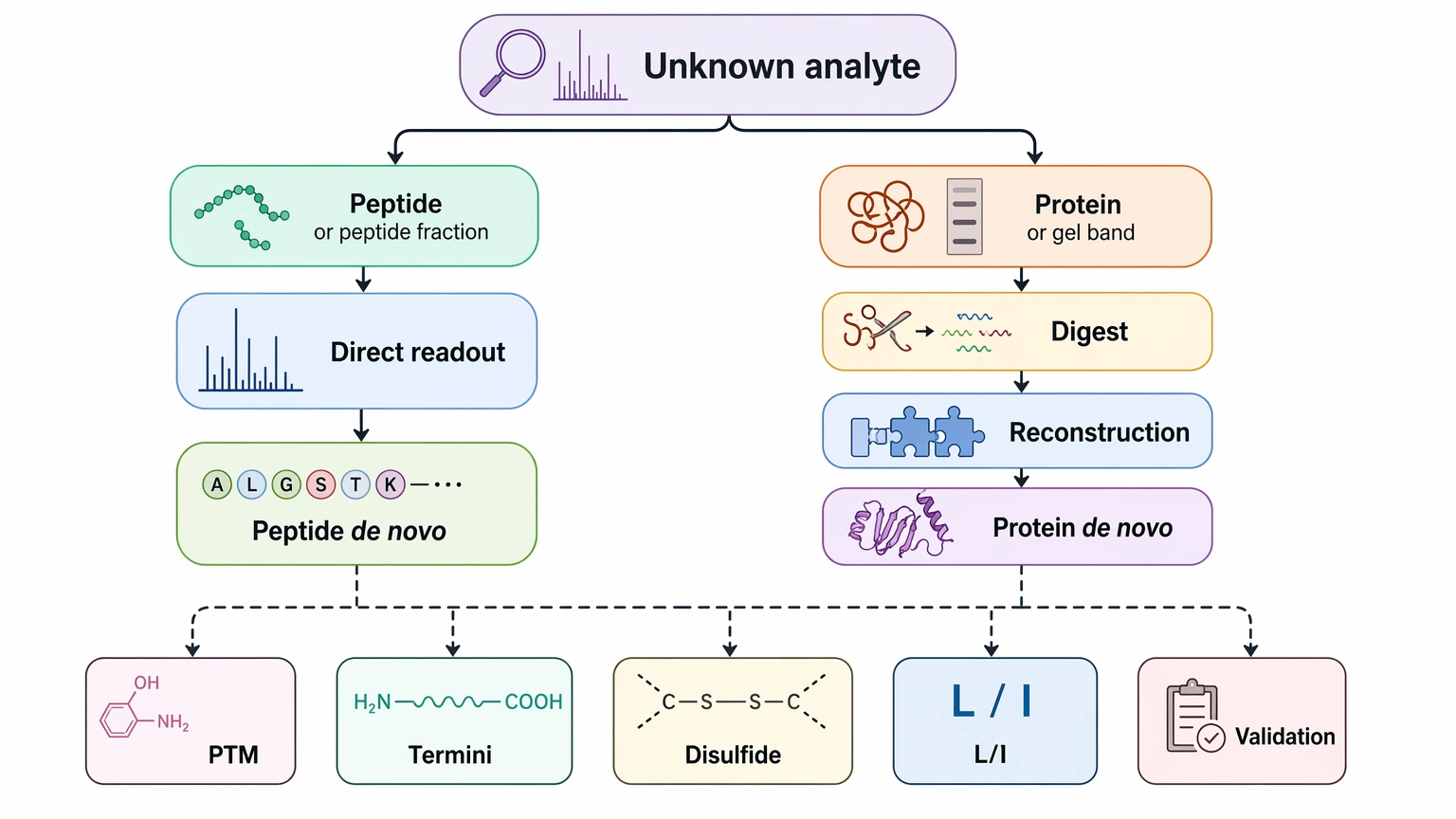
If the unknown analyte is already a peptide or a peptide-enriched fraction, the project usually fits peptide de novo sequencing. If the starting material is an intact protein, gel band, biologic candidate, or another protein sample that first has to be converted into overlapping peptide evidence, the project is usually better planned as protein de novo sequencing. That single choice changes the LC-MS/MS workflow, the burden of sequence assembly, the ambiguity you are likely to face, and the type of report that can be defended scientifically.
Quick Decision Guide
Choose peptide de novo sequencing when the interpreted unit is the peptide itself.
Choose protein de novo sequencing when protein-level conclusions must be reconstructed from digested peptide evidence.
Expect more direct readout for purified peptides and more layered confidence annotation for proteins.
Plan validation early when exact termini, post-translational modification (PTM) placement, disulfides, or leucine/isoleucine identity will affect the next decision.
Figure 1. Sample-Type Decision Path for De Novo Sequencing.
This is not just a size issue. In peptide de novo sequencing, the MS/MS spectrum often supports residue-by-residue inference from one analyte. In protein de novo sequencing, tandem mass spectrometry still generates peptide-level spectra, but those spectra then have to be organized into a larger protein conclusion. That difference shifts sample preparation, fragmentation priorities, confidence annotation, and reporting depth.
Where the Choice Usually Becomes Important
The distinction usually matters once a database search limitation becomes obvious. A venom fraction may contain a novel peptide with no useful reference. A synthetic peptide may include terminal blocking or truncation. A purified enzyme band may point only to a protein family rather than one exact sequence. A recombinant variant may contain substitutions or heterogeneous PTMs that prevent clean reference matching.
At that point, the practical question is not simply whether mass spectrometry can detect the analyte. The real question is what level of inference is needed: direct peptide sequence readout, a defensible sequence tag, or protein-level sequence assembly from multiple peptides. If you are comparing service routes at this stage, submit your requirements early so the workflow is built around the sample state and the readout you actually need.
Side-by-Side Comparison of the Two Workflows
The table below gives a practical starting point.
Sample type
Best fit
Main constraint
Next planning step
Purified unknown peptide
Peptide de novo sequencing
Blocked termini or dense modification can interrupt fragment interpretation
Confirm purity, amount, and expected PTM burden
Peptide mixture from venom, secretion, or hydrolysate
Peptide de novo sequencing
Co-eluting species can reduce spectral quality
Consider fractionation or targeted isolation
Intact purified protein
Protein de novo sequencing
Requires proteolytic digestion before informative de novo interpretation
Define enzyme strategy
Gel band or partially purified protein
Protein de novo sequencing
Background proteins may reduce sequence coverage
Improve cleanup and assess material recovery
Biologic variant or natural-source protein
Protein de novo sequencing
Mixed proteoforms complicate assembly
Combine intact protein vs digested peptide evidence
Takeaway: choose based on the starting analyte state and the intended level of sequence conclusion, not only on molecular mass.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
Workflow Entry: Direct Peptide Readout Versus Protein Reconstruction
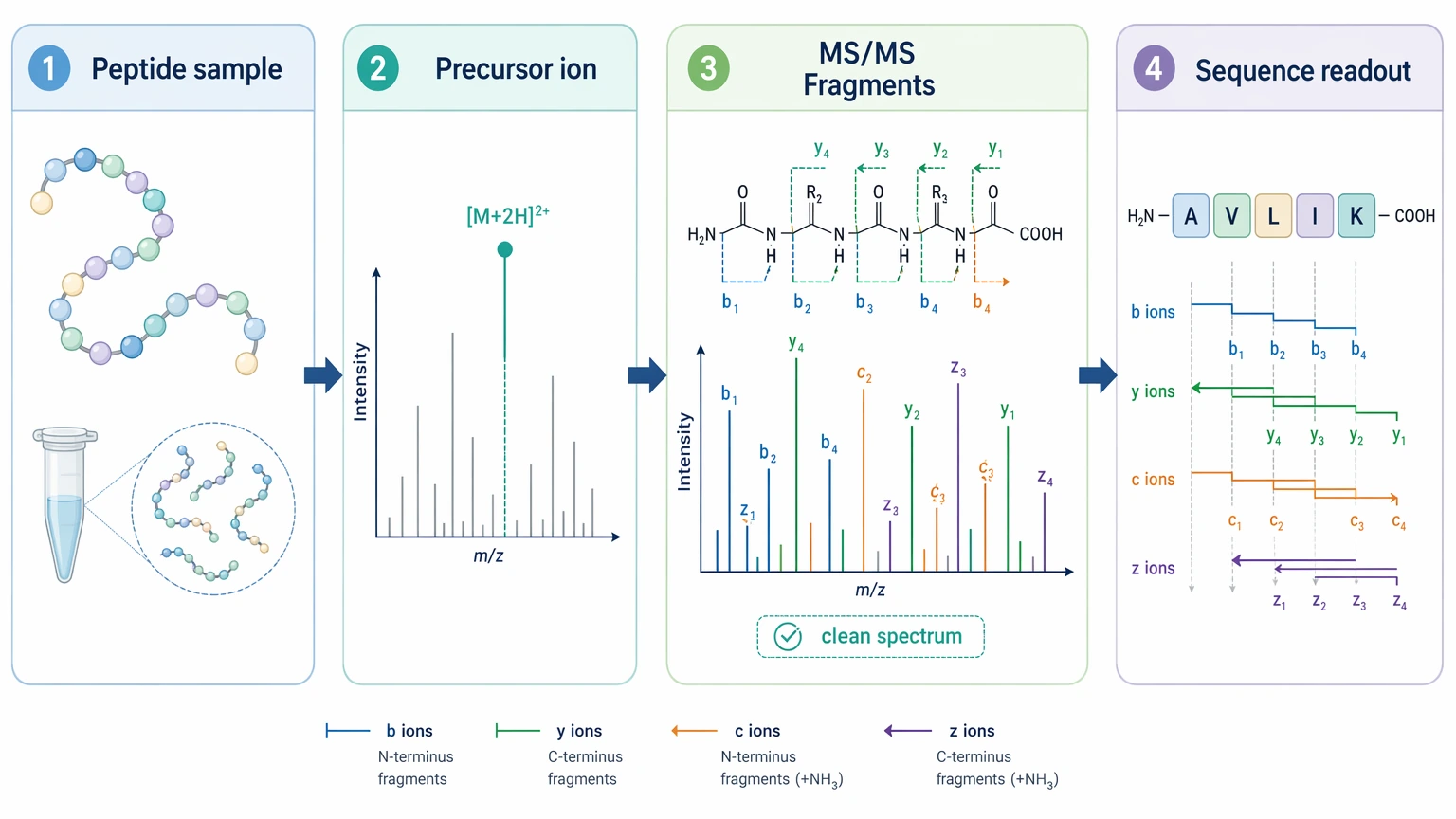
A peptide project may enter LC-MS/MS with little or no digestion. The main task is to interpret one precursor ion and its fragment ion series, often with emphasis on b ions / y ions and, in some settings, c ions / z ions. When the peptide is reasonably pure and fragments well, the analytical path can be fairly direct.
Figure 2. Peptide LC-MS/MS Readout Path.
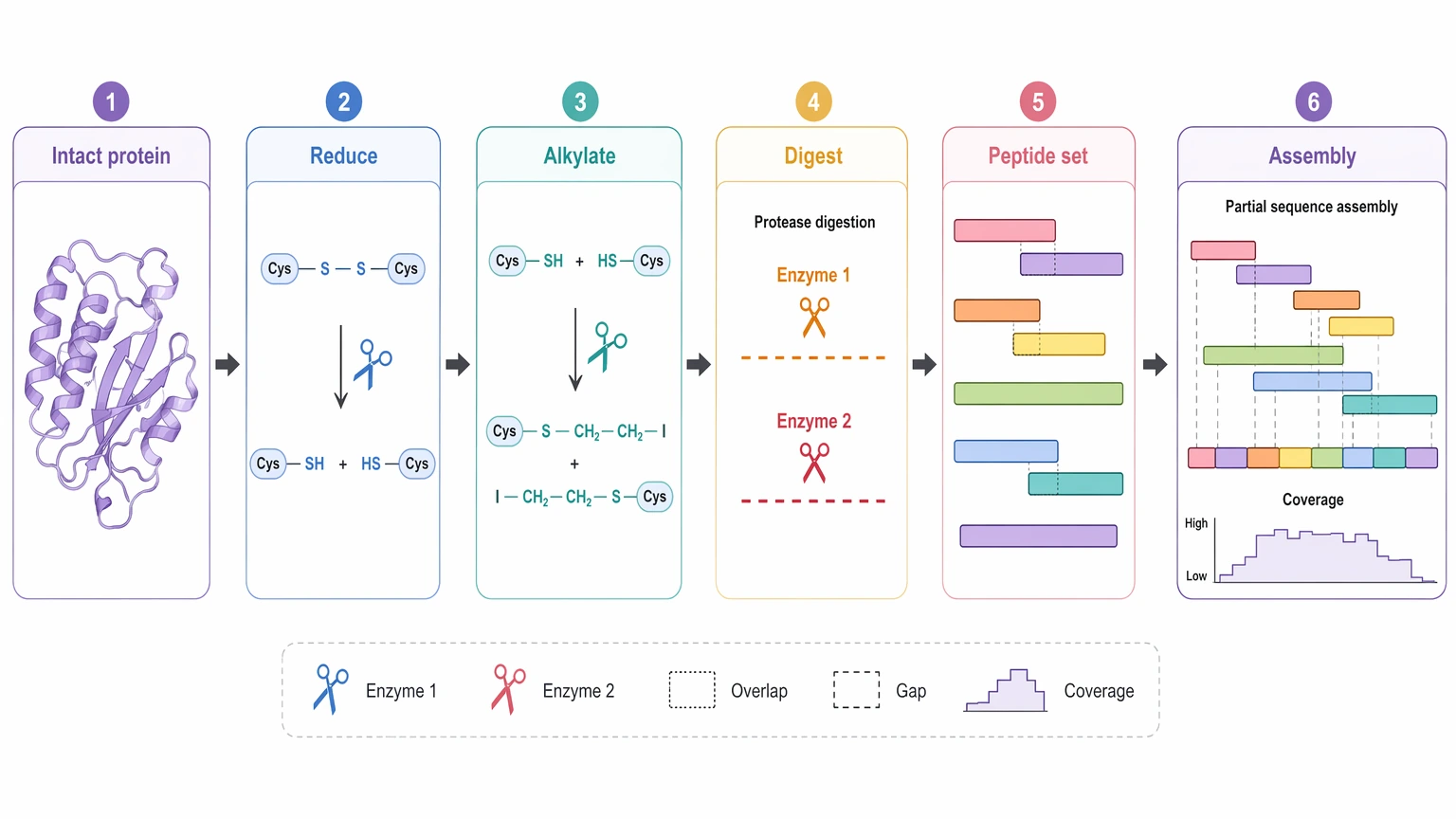
A protein project begins differently. The intact protein usually does not yield a complete answer through direct top-level interpretation alone. Instead, the sample is commonly reduced, alkylated, digested, and then analyzed as a peptide set. Those peptides become the evidence base for protein reconstruction. In that setting, digestion is part of the sequencing logic, not just a routine preparation step. One enzyme may leave gaps, while multi-enzyme digestion can create overlapping evidence that strengthens sequence assembly.
Figure 3. Protein Digest-to-Assembly Workflow for protein de novo sequencing depends on digest-derived overlapping peptide evidence for assembly.
How Sample Preparation and Fragmentation Shift the Logic
For peptide de novo sequencing, the main technical question is whether the peptide produces a fragment pattern rich enough for confident local interpretation. PTMs, blocked termini, and low-abundance neighboring species may interrupt ladder continuity, but the project still centers on one peptide backbone at a time.
For protein de novo sequencing, fragmentation quality still matters at the peptide level, but a second challenge appears: how well the peptide set connects into a larger model. Useful peptide spectra do not automatically produce a complete protein sequence. Gaps can remain because of uneven digestion, poor ionization of some regions, nonunique peptides, or incomplete overlap between peptide segments.
This also changes how PTMs are treated. In peptide projects, a PTM usually affects one analyte directly. In protein projects, PTMs may vary across a population of peptides and proteoforms. That means a modification can complicate both local interpretation and broader assembly.
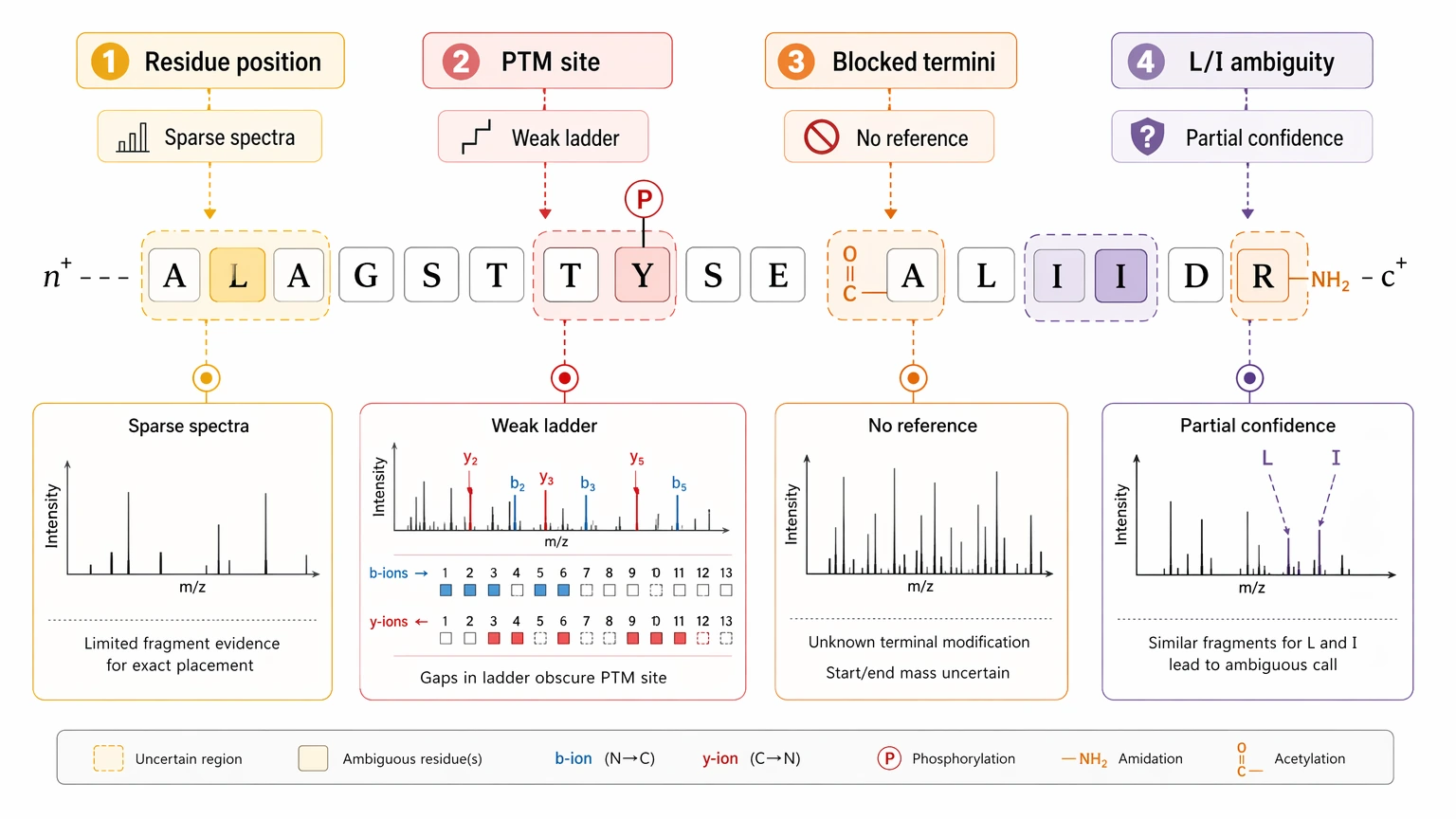
A practical limitation should be stated plainly: MS/MS-based de novo sequencing may leave unresolved residue positions, PTM localization uncertainty, or incomplete sequence confidence, especially when spectra are sparse, termini are blocked, or no database-supported context exists. This is especially relevant for leucine/isoleucine ambiguity, which often remains unresolved by routine fragmentation alone.
Figure 4. De Novo Sequencing Ambiguity Localization Map.
What the Readout Usually Looks Like
Readers often expect one “final sequence,” but the realistic deliverable depends on sample type.
Evidence type
Typical support
Common limitation
Typical follow-up
Continuous ladder from one peptide precursor ion
Strong peptide-level proposal
May retain isobaric residue ambiguity
Orthogonal residue confirmation if required
Distinct terminal fragment ion evidence
N- or C-terminal support
Blocked ends can weaken certainty
Targeted terminal confirmation
Overlapping peptide sequence tags from one protein digest
Regional protein assembly
Gaps remain without overlap
Add orthogonal digestion
PTM-bearing fragments
Local PTM annotation
Site placement may remain partial
Targeted site confirmation
Intact mass aligned with peptide evidence
Proteoform plausibility check
Does not prove full residue order
Use as supporting evidence
A peptide-focused report may provide a high-confidence candidate sequence, local PTM notes, and ambiguity flags at specific positions. A protein-focused report usually needs more layered reporting: region-by-region confidence annotation, peptide support mapping, PTM notes, and a clear separation between confirmed and inferred sequence regions.
Expected Results and Validation Methods
For peptide de novo sequencing, the immediate deliverable is often a candidate peptide sequence, one or more sequence tags, precursor and fragment evidence, and confidence notes for uncertain positions or termini. For protein de novo sequencing, the immediate deliverable is more often a structured package of peptide-level calls, regional assembly results, sequence coverage summary, PTM annotations, and a statement of which regions remain ambiguous.
That immediate output is not the same as follow-up confirmation. Follow-up validation is usually needed when a downstream decision depends on an exact residue call, terminal structure, disulfide connectivity, a regulatory-facing claim, or a site-specific PTM assignment. Practical confirmation routes may include targeted LC-MS/MS reanalysis, orthogonal digestion, intact mass correlation, terminal sequencing, or another residue-discriminating method.
If your project must separate peptide-level certainty from protein-level reconstruction before assay design or sequence handoff, contact MtoZ Biolabs to evaluate your project and align the report format with the actual validation burden instead of treating all de novo outputs as equally final.
Key Cautions and Practical Limits
Several limits matter more than broad workflow checklists.
Sample quality and amount limits: low recovery, mixed backgrounds, or very limited material can restrict repeat injections, alternate fragmentation, or parallel digests. This matters most for protein projects, where overlap is often necessary.
Controls and repeat expectations: one informative run may be enough for some clean peptide projects, but proteins more often benefit from repeat acquisition or orthogonal digestion to test consistency across peptide evidence.
Batch and contamination risk: gel bands, natural extracts, and partially purified proteins may carry background peptides that complicate assembly. Mixed samples can also produce misleading local sequence calls if co-isolated species contribute fragments.
Interpretation boundaries: a convincing local peptide call does not always justify a protein-level conclusion. In the same way, intact mass agreement supports plausibility but does not prove residue order.
When another method is the better next step: if exact L/I assignment, terminal blocking chemistry, disulfide pairing, dense glycosylation, or highly heterogeneous proteoforms drive the decision, another confirmatory method or outside sequencing support may be a better next step than repeated de novo interpretation alone.
A Practical Selection Framework
Choose peptide de novo sequencing when the sample is already peptide-sized, the decision unit is the peptide itself, and the main questions involve backbone sequence, truncation, or a local PTM pattern. Choose protein de novo sequencing when the starting material is a protein and the final conclusion must be assembled from digest-derived peptide evidence across multiple regions.
That rule is most useful for peptide discovery teams, assay developers, and analytical scientists working with novel peptides, gel bands, protein variants, biologic candidates, or modification-rich samples where reference matching is not enough. Before consultation, prepare the sample type, approximate amount, purity level, suspected PTMs, desired readout, and whether exact residue identity will affect the next experiment. With that information, you can evaluate your project, compare workflow options, and contact MtoZ Biolabs for a sequencing discussion that matches the real sample and the level of confidence you need.
FAQ
Can the same project involve both peptide and protein de novo sequencing?
Yes. Protein projects often include peptide-level de novo interpretation as one component, but the project is still classified by the final decision level. If the goal is protein reconstruction, peptide calls are supporting evidence rather than the endpoint.
Why can a protein project return only regional sequence information?
Because protein conclusions depend on overlapping peptide evidence. If some regions do not digest well, ionize poorly, or produce nonunique fragments, the report may support only certain sequence regions with confidence.
Does ETD always improve de novo sequencing results?
No. ETD or related methods can help in some peptide contexts, especially when labile PTMs or charge state support that chemistry, but they do not automatically fix sparse fragmentation or assembly gaps.
When is a sequence tag more useful than a forced full-sequence call?
A sequence tag is often the better output when one region is strong but other positions remain ambiguous. It can still guide targeted validation, reference screening, or family-level interpretation without overstating certainty.
How should I prepare a sample for an early project-fit discussion?
Bring the sample identity state, concentration or estimated amount, purification history, suspected modifications, any prior LC-MS/MS results, and a clear statement of what downstream choice depends on the sequence result.