





Monoclonal Antibody Sequencing or De Novo Sequencing: How to Choose Based on Starting Material
- the exact starting material type
- approximate amount or concentration available
- whether the sample is purified antibody, hybridoma, recombinant construct, or archival material
- whether VH and VL recovery or only CDR confirmation is needed
- any known isotype or subclass context
- any previous RACE, NGS, Sanger, or LC-MS/MS data
- whether the sample is degraded, old, reformulated, or inconsistently labeled
- whether the final output must support recombinant re-expression
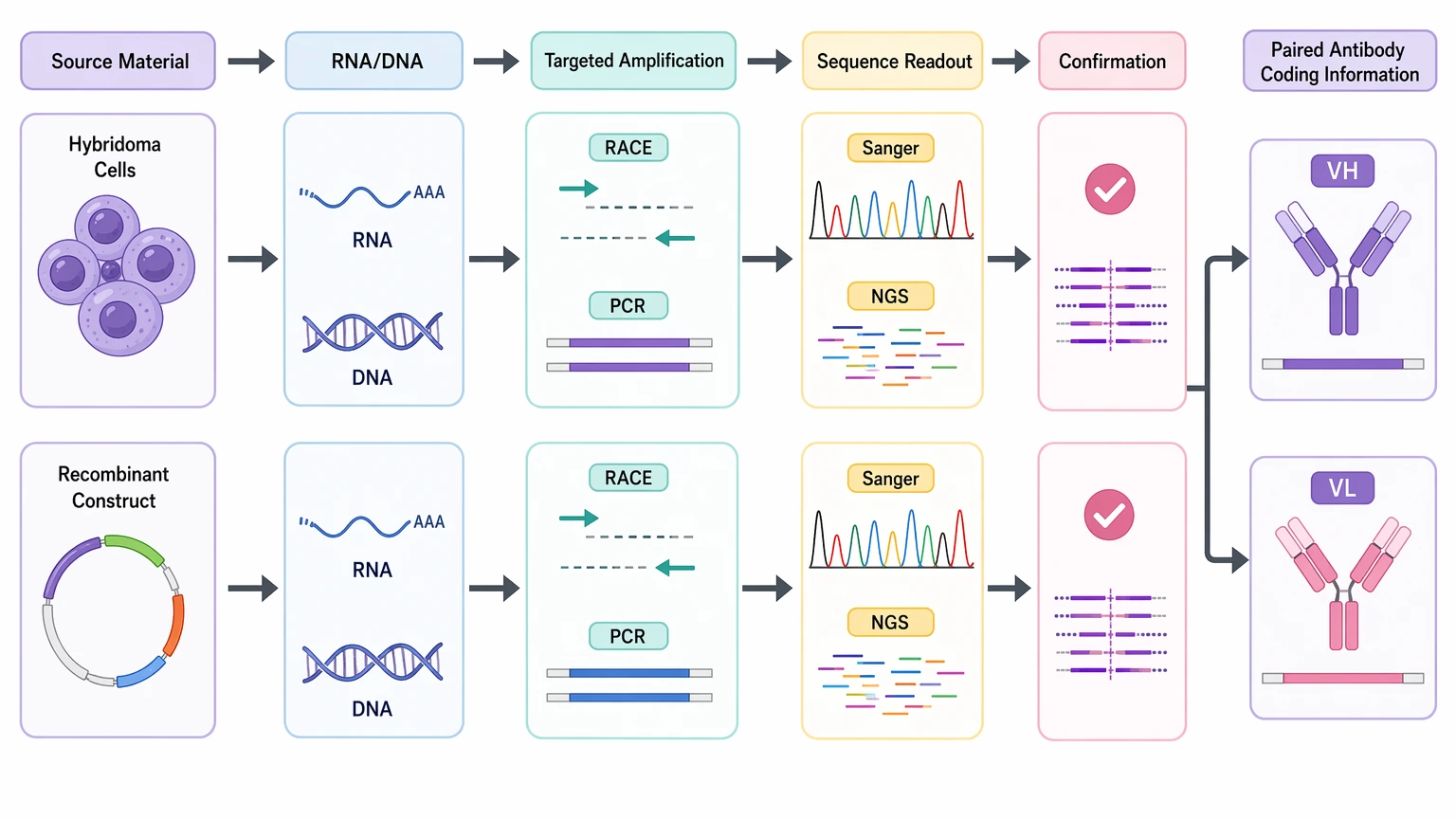
Monoclonal antibody sequencing usually begins with cellular or DNA-derived material when a hybridoma or recombinant construct is still on hand, because those inputs allow more direct recovery of paired VH and VL coding information. De novo antibody sequencing is usually the better place to start when purified antibody is the only material left, especially for legacy clone rescue or archival recovery when no usable nucleic acid source remains.
The real decision is not which service label sounds more sophisticated. It is which starting material can still yield a usable heavy chain and light chain sequence with enough confidence for the job you actually need to do. If the goal is CDR confirmation from a purified antibody lot, LC-MS/MS-based sequence recovery may be enough. If the goal is a full-length sequence for recombinant re-expression, transcript-based sequencing from a hybridoma or direct recovery from a recombinant construct is often the cleaner path.
Start with the material you actually have
This choice usually comes up after the antibody already exists but the sequence record does not. A team may have a purified antibody vial from an older program, a frozen hybridoma pellet, a recombinant construct with incomplete annotations, or archival material with uncertain chain records. At that stage, the project is no longer about discovery. It is about recovering a usable variable region sequence from whatever source still remains.
A poor route choice early on can consume limited sample and still leave the main questions unresolved. Protein-only workflows may use up valuable purified antibody and still leave ambiguity in low-coverage regions. Transcript-based sequencing is often more direct, but degraded RNA, mixed hybridoma backgrounds, or incomplete construct files can limit recovery. The most practical comparison therefore comes down to four questions: what sequence can be recovered, how confident chain pairing will be, what ambiguity is likely to remain, and how much validation is needed before recombinant re-expression.
Two routes, two sequence sources
Monoclonal antibody sequencing here refers to sequence recovery from biologic source material such as hybridoma cells or a recombinant construct. Because the sequence comes from RNA or DNA, these workflows can often recover the antibody coding sequence more directly. Common methods include transcript-based sequencing, RACE, targeted PCR, Sanger confirmation, and NGS.
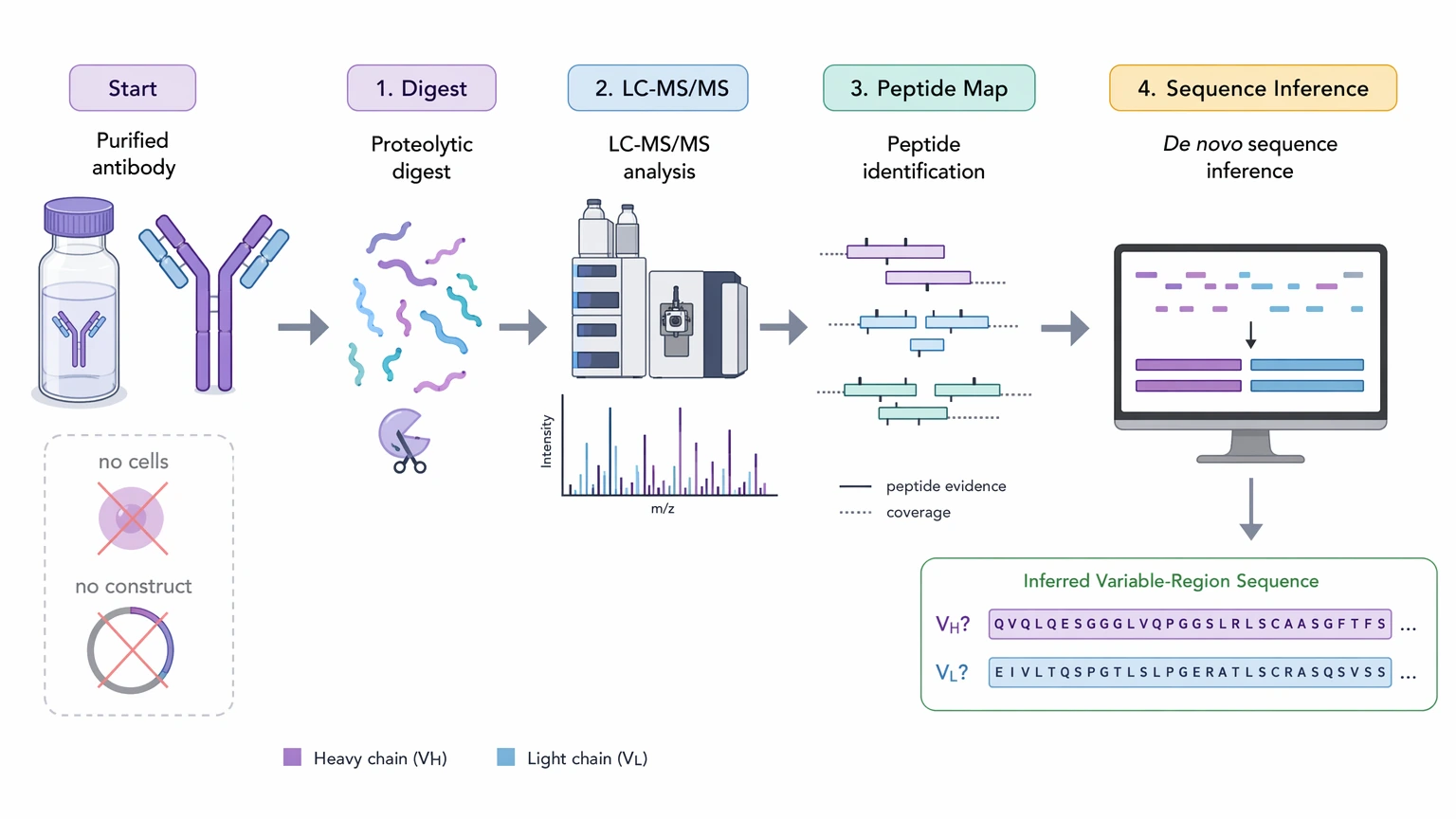
De novo antibody sequencing refers to protein-level sequence inference from purified antibody, usually through LC-MS/MS and peptide mapping. This route matters when no viable cells or reliable construct records remain. The output can be highly informative, but it is reconstructed from peptide evidence rather than read directly from a nucleic acid template.
Four comparison points that matter most
Recoverable sequence scope
The first question is what the project needs to recover. Some teams only need CDR confirmation. Others need the full variable region, or a full-length sequence that can be used for vector design. Hybridoma and recombinant construct workflows more often support complete VH and VL recovery. Purified antibody can also support broad sequence recovery, but the outcome depends on peptide coverage across the variable region and on how clearly the peptide evidence supports each residue call.
Chain pairing confidence
A useful result is not just a list of sequence fragments. It needs to support correct heavy chain and light chain assignment, ideally with matched VH and VL for downstream work. Hybridoma and construct-derived workflows usually provide stronger chain pairing confidence than protein-only inference. That difference matters even more when the end goal is recombinant re-expression rather than basic identity checking.
Sequence ambiguity
Protein-level workflows can leave leucine/isoleucine ambiguity because those residues are isobaric in many LC-MS/MS settings. Post-translational modifications, glycosylation, and incomplete peptide mapping can add further uncertainty to sequence interpretation. Nucleic-acid-based workflows avoid some of those issues, but they come with their own failure modes, including degraded RNA, mixed transcripts, and nonproductive rearrangements.
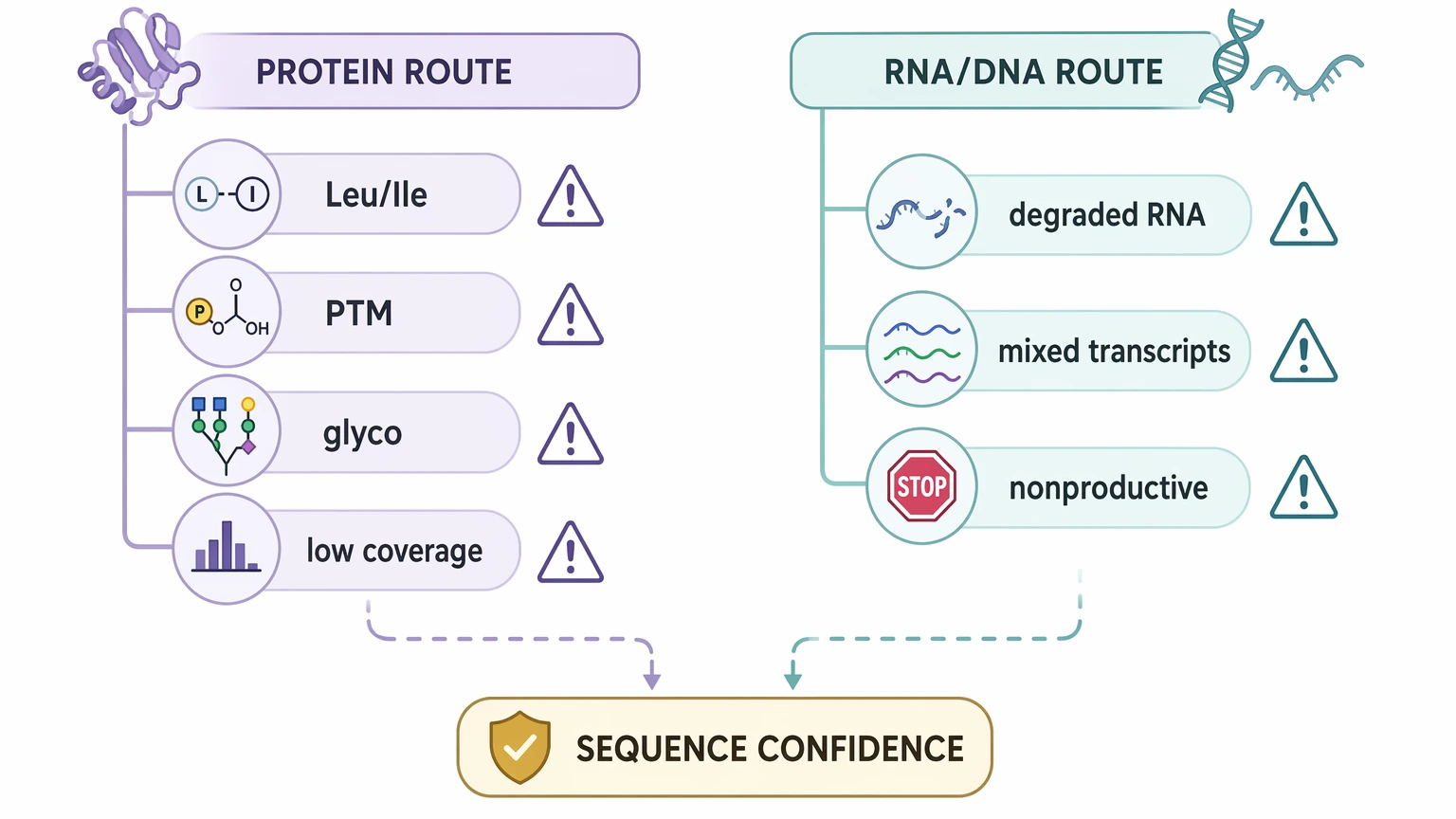
Validation burden
A final sequence package may be a direct coding sequence, a ranked set of candidate calls, or a protein-inferred sequence that still needs orthogonal confirmation. The more the project depends on recombinant re-expression, the more the validation plan matters. That plan may include transcript confirmation, recombinant expression checks, or protein-level comparison to the original material.
Starting-material decision matrix
A starting-material matrix is often the quickest way to match the project to the right route.
| Starting material | Recommended first route | Recoverable sequence scope | Chain pairing confidence | Common risk or ambiguity | Re-expression readiness |
|---|---|---|---|---|---|
| Purified antibody only | De novo antibody sequencing | CDR to variable region, depending on coverage | Moderate | Leucine/isoleucine ambiguity, incomplete coverage, PTM interference | Usually needs additional confirmation |
| Hybridoma available | Monoclonal antibody sequencing by transcript-based sequencing | Full VH and VL, often with coding-sequence context | High if clone integrity is preserved | RNA degradation, mixed transcripts, background rearrangements | Often well suited |
| Recombinant construct available | Monoclonal antibody sequencing from construct-derived DNA | Full insert recovery if both chains are present | High | Missing chain records, version drift, incomplete annotations | Often strong if sequence mapping is verified |
| Archival material | Triage first, then choose the strongest surviving source | Variable | Variable | Degradation, uncertain provenance, low input, chain mismatch risk | Often requires staged validation |
How the choice changes by sample scenario
Purified antibody only
If purified antibody is the only available starting material, de novo antibody sequencing is usually the most realistic entry point. This is especially relevant for older antibody lots, rescued repository samples, and archival materials that outlasted their hybridoma or construct records. It can support CDR analysis and substantial variable region sequence recovery.
Its main limitations come back to evidence quality. Sequence recovery depends on peptide mapping depth, coverage across the variable region, and how interpretable modified peptides are. A de novo result may be strong enough to move the project forward, but it often includes explicit sequence ambiguity annotations. For recombinant re-expression, teams should expect some degree of orthogonal confirmation rather than assume every residue call is final.
Hybridoma available
If a hybridoma still exists, monoclonal antibody sequencing by transcript-based sequencing is usually the preferred route for full VH and VL recovery. It offers more direct access to the heavy chain and light chain coding sequence and usually gives higher chain pairing confidence than protein-only inference.
That advantage is greatest when the cells are still intact and the clone history is reasonably controlled. Hybridoma availability does not automatically guarantee a clean result. Older or mixed cultures may contain background transcripts, and degraded RNA can weaken recovery. Even so, when recombinant re-expression is the goal, hybridoma-derived sequence recovery is often the clearest place to start.
Recombinant construct available
A recombinant construct changes the decision logic again. If plasmid DNA or a construct archive is still available, sequence recovery may be less about discovery and more about confirming what is actually present in the insert. In those cases, monoclonal antibody sequencing from construct-derived material is often the shortest route to a usable coding sequence.
The main risk is usually not chemistry. It is documentation quality. Older programs may have partial maps, mislabeled versions, or only one documented chain. If both heavy chain and light chain inserts can be verified, construct-derived recovery is usually more direct than de novo antibody sequencing from purified protein.
Archival or incomplete material
Archival material is the hardest category because it may contain several weak information sources at once: a partially degraded pellet, an old purified antibody vial, incomplete construct files, or uncertain sample labels. Here, the first step is triage rather than immediate route selection. Check whether confirmed construct DNA exists, whether transcript-based sequencing is still realistic, and whether the purified antibody is intact enough for LC-MS/MS.
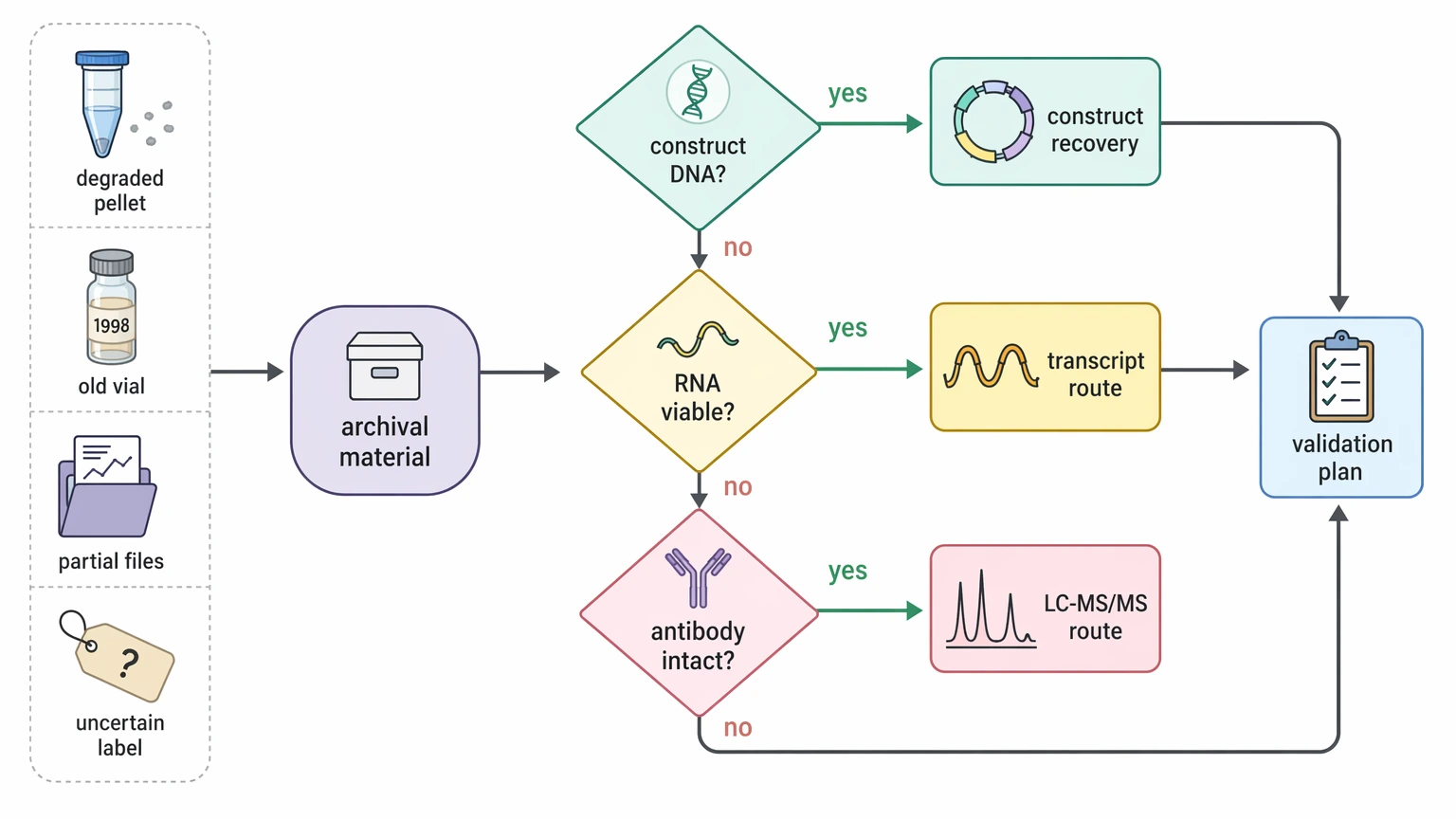
When sample is scarce, route selection should also avoid unnecessary consumption. If your archive includes mixed or degraded materials and the workable path is still unclear, you can submit your requirements and ask MtoZ Biolabs to evaluate the project around surviving sample type, likely sequence ambiguity, and the validation plan before full sequencing begins.
Quick decision framework
If you have purified antibody only, start with de novo antibody sequencing.
If you have a hybridoma, start with transcript-based monoclonal antibody sequencing unless RNA quality is clearly compromised.
If you have a recombinant construct, verify the insert first and use construct-derived sequence recovery as the primary path.
If you have both protein and cellular material, use the nucleic-acid-derived route for primary sequence recovery and keep the purified antibody as an orthogonal confirmation layer.
If you are trying to rescue a legacy antibody for recombinant re-expression, prioritize the route that offers the strongest chain pairing confidence and the least unresolved sequence ambiguity, even if that route is not the one your team uses most often.
At the project-selection stage, a concise intake package helps. Include the starting material category, available amount, expected integrity, whether heavy chain and light chain records are missing or partial, whether the goal is CDR confirmation or full-length sequence recovery, and whether recombinant re-expression is planned. With that information, MtoZ Biolabs can evaluate your project and help match the workflow to the material before scarce sample is committed.
What to prepare before consultation
A useful submission usually includes:
These details matter because they shape deliverable confidence, chain pairing strategy, and the validation plan.
Comparison summary and next step
For starting-material-based decisions, the main rule is straightforward: use monoclonal antibody sequencing when a hybridoma or recombinant construct still provides access to the coding source, and use de novo antibody sequencing when purified antibody is the only realistic substrate left for sequence recovery. The tradeoff is practical rather than abstract: direct coding-sequence access versus protein-level inference, with chain pairing, sequence ambiguity, and validation burden determining how ready the output is for downstream use. If your project involves a purified legacy lot, a surviving hybridoma, a partially annotated construct, or mixed archival material, prepare those sample details and contact MtoZ Biolabs to discuss the workflow, validation needs, and project fit before moving into full sequence recovery.
FAQ
Can de novo antibody sequencing distinguish leucine from isoleucine in every position?
Not always. LC-MS/MS evidence may leave leucine/isoleucine ambiguity in some peptides, so the report may include candidate residue calls or recommend orthogonal confirmation for positions that affect downstream cloning.
What if the hybridoma and purified antibody appear to disagree?
That can happen if the archived protein lot and the surviving cells do not come from the same passage history or if one source is mislabeled. In that situation, comparing transcript-based sequencing with protein-level peptide mapping can help clarify whether the discrepancy is biological, archival, or analytical.
Is a recombinant construct always enough to recover both chains?
Only if both inserts are actually present and correctly mapped. Some archives preserve one chain clearly but lose the second chain record, so the construct should be checked for completeness before it is treated as a full sequence source.
When is archival material still worth testing?
Archival material is still useful when at least one layer of information remains interpretable, such as recoverable plasmid DNA, amplifiable RNA, or a sufficiently intact purified antibody lot. The key question is not age by itself but what sequence evidence still survives.
What should a deliverable confidence statement include?
A useful confidence statement should describe sequence scope, chain pairing support, unresolved ambiguities, evidence type, and any recommended validation steps. That makes the result easier to judge for identity confirmation versus recombinant re-expression planning.
How to order?

