





Mate Pair Sequencing for De Novo Assembly: When Long-Insert Libraries Still Improve Scaffold Continuity
- longer connected residue stretches across the backbone
- fewer candidate assemblies that explain the same LC-MS/MS evidence
- improved confidence at the N-terminus and C-terminus
- narrower residue-order ambiguity in low-coverage regions
- more defensible PTM localization
- better agreement between bottom-up interpretations and intact mass constraints
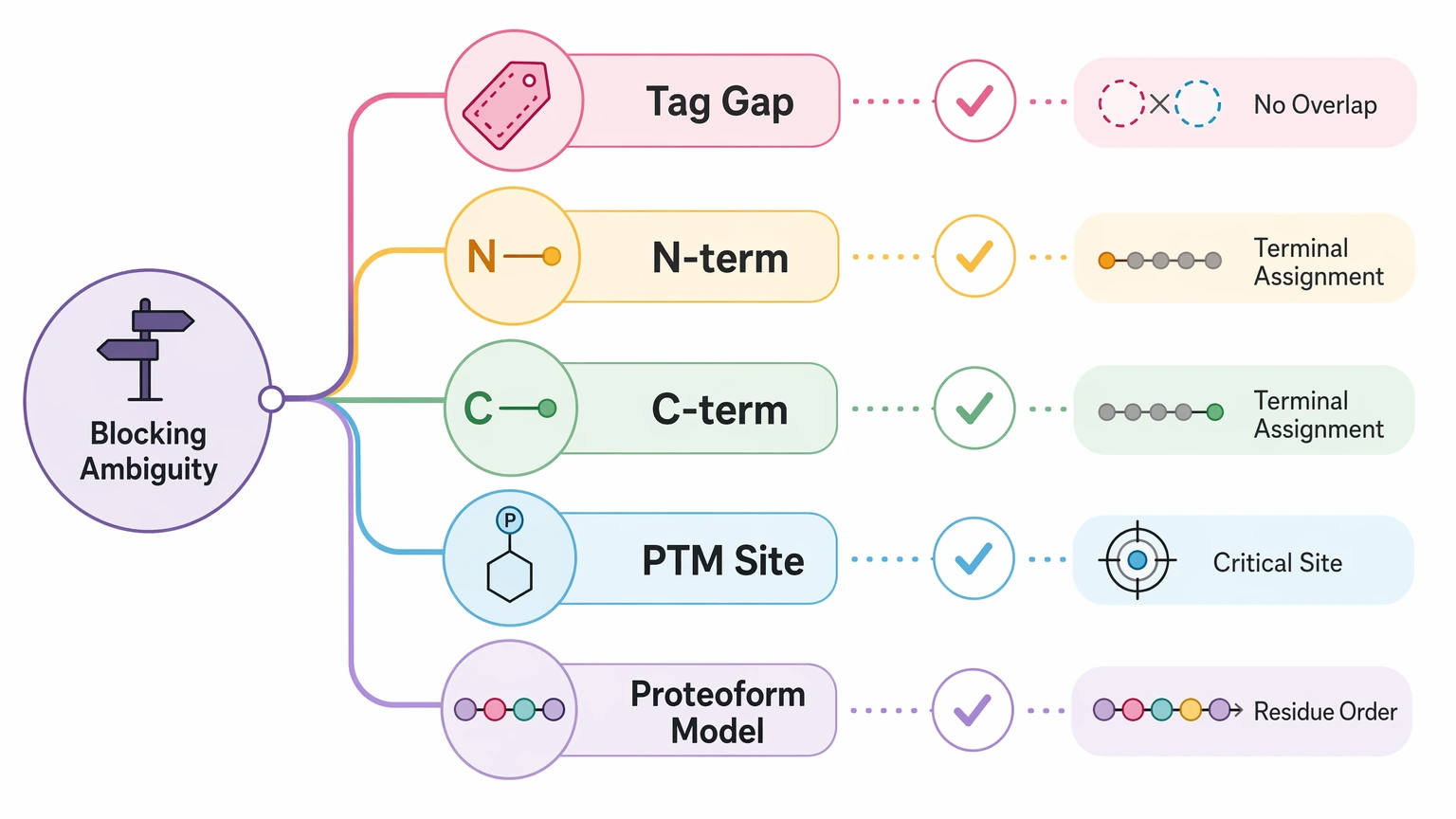
- two strong sequence tag regions that do not overlap
- uncertain N-terminus or C-terminus assignment
- unresolved PTM localization on a decision-critical site
- multiple proteoform models with the same approximate intact mass
- repeated local tags that do not settle residue order
- Complementary protease digestion helps most when one digest leaves unbridged gaps. New cleavage patterns can create overlapping tags that improve sequence coverage and reduce residue-order ambiguity.
- ETD, ECD, or sometimes UVPD can help when PTM retention or alternate fragmentation behavior matters more than raw scan count.
- Intact mass and top-down proteomics become more useful when the main question is whether peptide-level pieces belong to one proteoform, a truncated form, or a mixture.
- Targeted follow-up fragmentation works best when only one or two decision-critical positions remain uncertain.
- an updated de novo peptide sequencing or de novo protein sequencing report
- ranked candidate assemblies with stated confidence boundaries
- overlap maps across complementary digests
- fragmentation-mode comparison for modified regions
- intact mass agreement or disagreement with candidate models
For partially resolved unknowns or PTM-rich analytes, the real question is not whether you can collect more LC-MS/MS data. It is whether a different type of evidence will remove the ambiguity that is still blocking the sequence call. When you already have strong local sequence tags but still cannot lock down residue order, PTM localization, terminal assignment, or proteoform separation, orthogonal evidence often does more than another round in the same acquisition mode.
Quick decision guide
Add orthogonal evidence now when you already have credible sequence tags but still cannot bridge overlaps, localize PTMs, define the N-terminus or C-terminus, or separate competing proteoform models.
Stay with the current workflow when the unresolved gaps do not change the project decision.
Switch methods instead of adding more similar spectra when the bottleneck is intact-level context, disulfide-related structure, or a persistent interpretation limit such as leucine/isoleucine ambiguity.
Why This Decision Matters in De Novo Work
The fixed title uses genome-assembly language, but the closest equivalent in de novo peptide sequencing and de novo protein sequencing is sequence continuity. In this setting, “scaffold continuity” means turning disconnected sequence-tag evidence into a coherent residue order with enough confidence to support a report, a design decision, or a follow-up experiment.
This becomes a practical problem when the first tandem mass spectrometry pass is informative but still incomplete. A team may already know the analyte is novel, truncated, engineered, or heavily modified. Even then, database-search support can stay weak because the target is missing from reference databases, carries unexpected PTMs, or diverges enough from known homologs that short local matches do not settle the full interpretation.
What Better Continuity Actually Means
A more continuous de novo result is not just a bigger data package. In practice, it usually means:
That distinction matters. Repeated short-fragment evidence can raise the spectral count without changing the answer. In de novo protein sequencing, the useful measure is ambiguity reduction, not data volume.
The Main Causes of Fragmented Output
For this decision, four cause categories usually matter most.
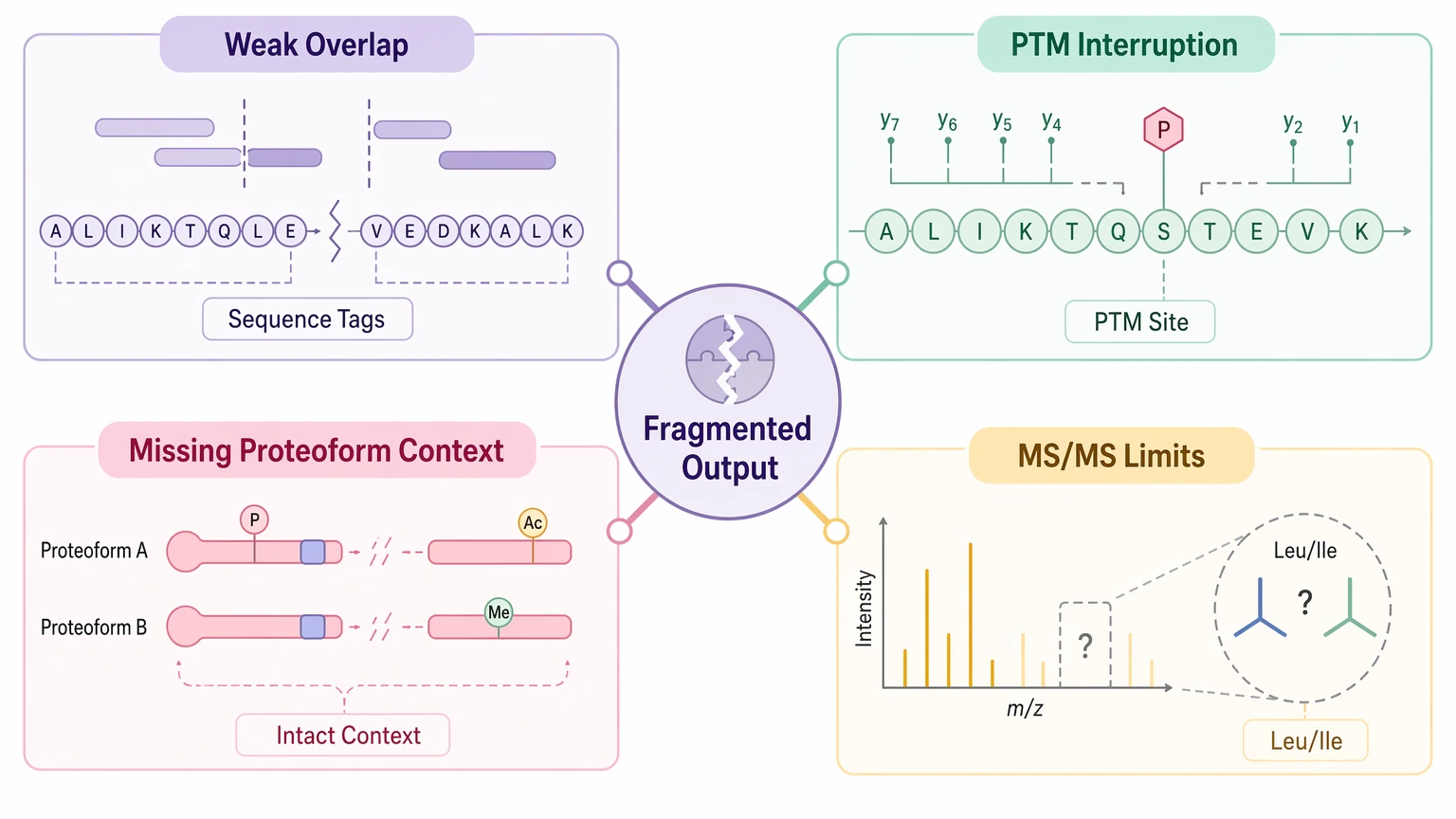
Weak overlap between otherwise good sequence tags
A single digest can produce accurate local tags and still leave large gaps between them. The same thing happens when one fragmentation mode keeps favoring certain sequence regions while missing others. In that situation, the workflow is generating real information, just not enough overlap to connect it.
PTM-driven interruption of backbone fragmentation
PTMs can disrupt ion-series continuity, especially when modifications are labile, clustered, or present across multiple forms of the analyte. HCD or CID may still yield useful sequence tags, but PTM localization and long contiguous interpretation can remain unstable.
Missing proteoform-level context
Bottom-up proteomics can leave you with several plausible assemblies at once when truncation, engineered changes, or mixed proteoforms are involved. Shared peptides may support more than one model, while the intact-molecule constraint is still absent.
Interpretation limits intrinsic to MS/MS
Some ambiguity is simply hard to close with more of the same data. Isobaric residues, especially leucine/isoleucine ambiguity, are a familiar example. PTM placement can also remain partly unresolved when fragment coverage is sparse around the modified site. Even after orthogonal evidence is added, tandem mass spectrometry may not assign every residue or PTM position with the same confidence.
A Method-Selection Workflow for Adding Long-Range Evidence
This is a method-selection problem, so the best way to handle it is to match the unresolved question to the smallest evidence expansion likely to change the decision.
Step 1: Define the exact ambiguity that still matters
Before adding another experiment, write down what is actually blocking the report. Useful examples include:
If resolving the remaining ambiguity would not change the project outcome, more expansion may add work without adding value.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
Step 2: Check whether the first-pass dataset is worth extending
Use the initial LC-MS/MS result as a gate. The current dataset should already contain credible de novo signal, not just noise or poorly fragmented precursors.
<div class="article-table-wrap" style="overflow-x:auto;margin:16px 0 20px 0;"> <table style="width:100%;border-collapse:collapse;table-layout:auto;font-size:14px;line-height:1.45;"> <thead> <tr> <th style="padding:10px 12px;text-align:left;background:#f3f6f8;border:1px solid #d9e2ec;font-weight:600;vertical-align:top;">Evidence</th> <th style="padding:10px 12px;text-align:left;background:#f3f6f8;border:1px solid #d9e2ec;font-weight:600;vertical-align:top;">What it supports</th> <th style="padding:10px 12px;text-align:left;background:#f3f6f8;border:1px solid #d9e2ec;font-weight:600;vertical-align:top;">Main limit</th> <th style="padding:10px 12px;text-align:left;background:#f3f6f8;border:1px solid #d9e2ec;font-weight:600;vertical-align:top;">Best next move</th> </tr> </thead> <tbody> <tr> <td style="padding:10px 12px;text-align:left;border:1px solid #e5edf3;vertical-align:top;">Several high-confidence sequence tag regions</td> <td style="padding:10px 12px;text-align:left;border:1px solid #e5edf3;vertical-align:top;">Real de novo signal is present</td> <td style="padding:10px 12px;text-align:left;border:1px solid #e5edf3;vertical-align:top;">Overlap may still be weak</td> <td style="padding:10px 12px;text-align:left;border:1px solid #e5edf3;vertical-align:top;">Add complementary protease digestion</td> </tr> <tr> <td style="padding:10px 12px;text-align:left;border:1px solid #e5edf3;vertical-align:top;">Useful b/y series but unstable modified-region interpretation</td> <td style="padding:10px 12px;text-align:left;border:1px solid #e5edf3;vertical-align:top;">Backbone fragmentation is partly informative</td> <td style="padding:10px 12px;text-align:left;border:1px solid #e5edf3;vertical-align:top;">PTM localization remains weak</td> <td style="padding:10px 12px;text-align:left;border:1px solid #e5edf3;vertical-align:top;">Add ETD or ECD</td> </tr> <tr> <td style="padding:10px 12px;text-align:left;border:1px solid #e5edf3;vertical-align:top;">Several bottom-up assemblies fit the same target</td> <td style="padding:10px 12px;text-align:left;border:1px solid #e5edf3;vertical-align:top;">Partial reconstruction exists</td> <td style="padding:10px 12px;text-align:left;border:1px solid #e5edf3;vertical-align:top;">Proteoform context is missing</td> <td style="padding:10px 12px;text-align:left;border:1px solid #e5edf3;vertical-align:top;">Add intact mass or top-down proteomics</td> </tr> </tbody> </table> </div>
The takeaway is straightforward: expand only when the current evidence is real, incomplete, and incomplete in a way that still matters to the project.
Step 3: Match the ambiguity type to the right evidence layer
Different add-ons answer different questions.
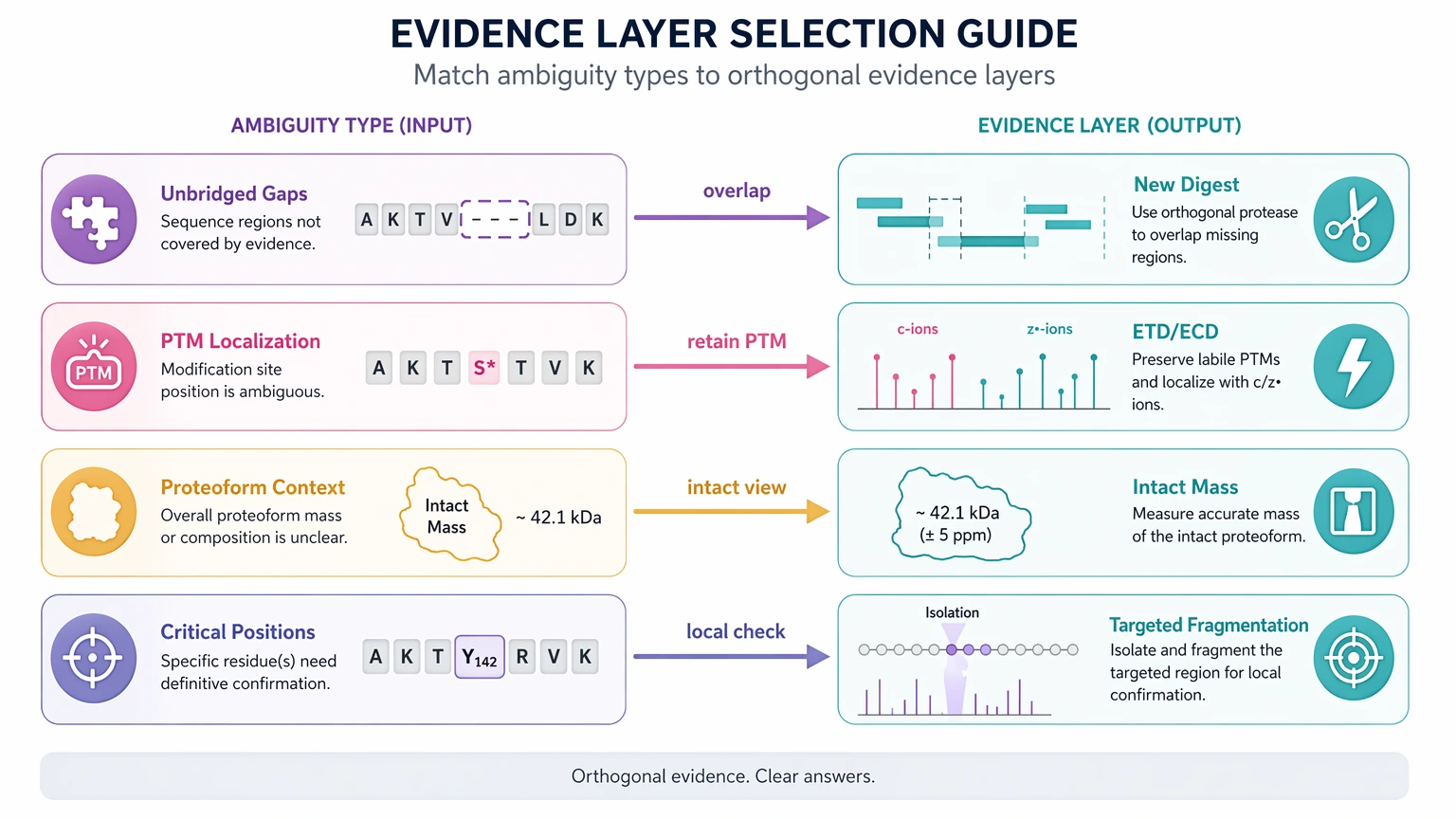
If your team is weighing these options before committing more sample, you can submit your requirements to MtoZ Biolabs to evaluate your project against the ambiguity type, sample state, and the likely report gain from each evidence layer.
Step 4: Decide whether the likely gain justifies the extra sample
Extra analysis makes sense when one nonredundant evidence layer could turn a fragmented interpretation into a usable sequence assignment. It makes less sense when the remaining gap is minor, the sample amount is already limiting, or the new experiment is likely to reproduce the same blind spots.
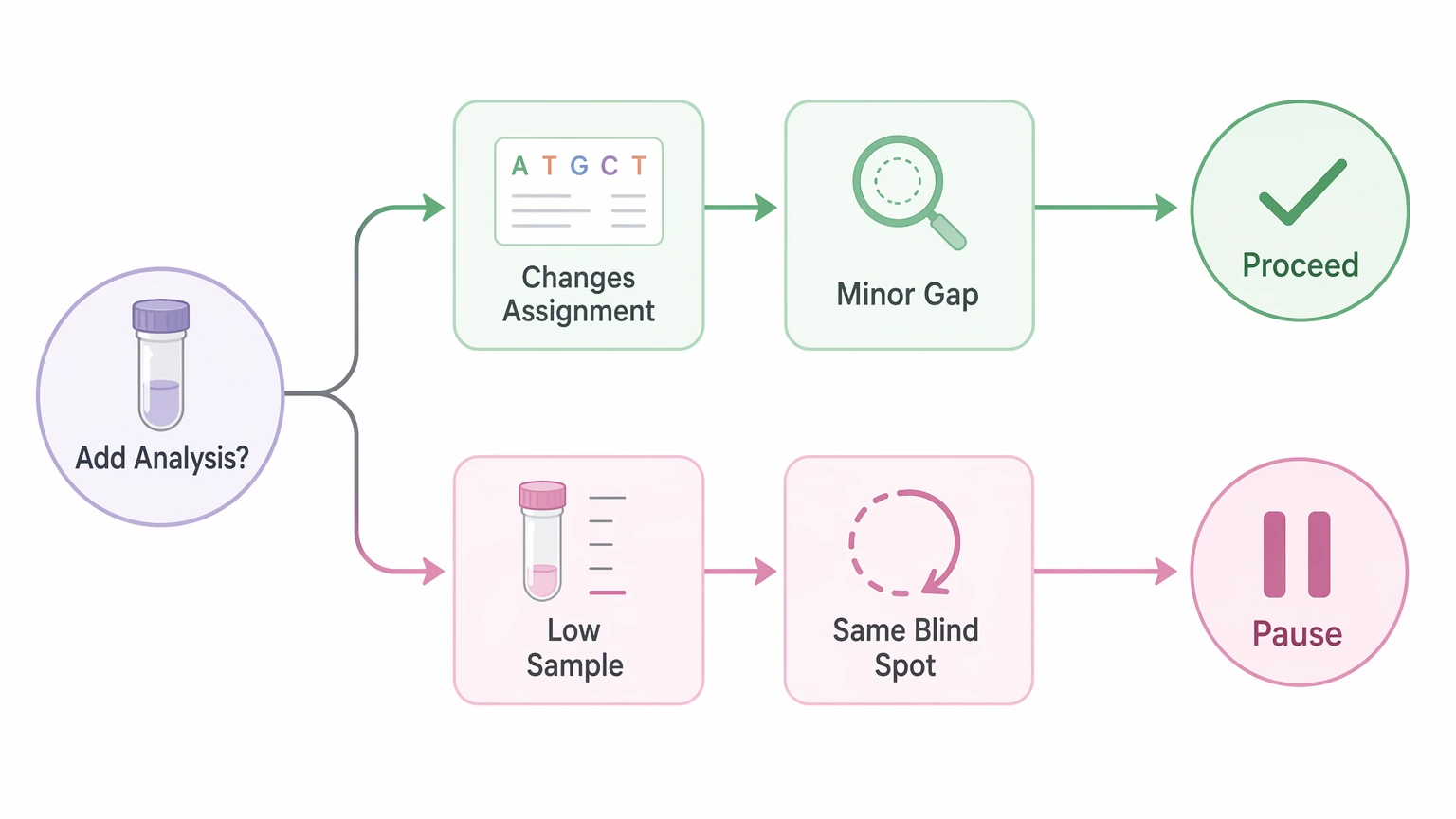
As a working rule, proceed when the unresolved issue changes sequence identity, PTM localization, or proteoform interpretation. Pause when the current result already supports the decision you need to make.
Expected Results and Validation Strategy
After a well-chosen expansion, the immediate deliverable should be a tighter interpretation, not a promise of full sequence closure. Common short-term gains include longer connected sequence tags, fewer incompatible assemblies, tighter PTM localization, and better agreement between assembled sequence logic and intact mass.
Follow-up confirmation is a separate step. Immediate deliverables can include:
Confirmation work should then test the claims most likely to affect downstream use. That may include targeted LC-MS/MS validation, disulfide bond mapping, orthogonal terminal analysis, or additional intact-level review. Validation strategy matters most when the final call involves novel proteins, site-specific PTM claims, or proteoform discrimination.
Key Cautions and Practical Limits
Sample quality and sample amount are the first hard limits. Complementary protease digestion, top-down proteomics, and targeted follow-up each consume material, so scarce or heterogeneous samples force real prioritization.
Controls and repeat expectations matter too. If PTM localization is the issue, repeating the same HCD workflow may add very little, while one orthogonal fragmentation mode may answer more. If contamination or carryover is possible, low-level foreign peptides can distort sequence-tag assembly and create false continuity.
Interpretation boundaries should stay explicit. Some reports will still include qualified statements about leucine/isoleucine ambiguity, partially localized PTMs, or multiple candidate proteoforms ranked by supporting evidence. That is not a workflow failure. It is an honest statement of sequence confidence.
In some cases, another method is the better next step. A purified protein with a terminal sequencing question may gain more from an alternate targeted approach than from repeated bottom-up expansion. If the project is stalled by sample limits or unusually complex modification patterns, contact MtoZ Biolabs to discuss the study, submit your requirements, and determine whether the next step should be orthogonal MS evidence, a different validation method, or a narrower decision-focused analysis.
Conclusion
In de novo peptide sequencing and de novo protein sequencing, the “mate pair” logic becomes useful when initial LC-MS/MS already gives you credible local tags but still falls short on sequence continuity, PTM localization, terminal assignment, or proteoform interpretation. The best expansion is usually the smallest orthogonal evidence layer that addresses the exact ambiguity, whether that means complementary protease digestion, alternate backbone fragmentation, intact mass support, or targeted confirmation. For novel, engineered, truncated, or PTM-rich analytes, that choice often decides whether the project ends with interesting fragments or with a report that is strong enough to guide the next experiment. If you are planning a difficult sequencing project, prepare the existing LC-MS/MS data, sample constraints, suspected modifications, and the exact report question so the workflow can be chosen with clear technical boundaries from the start.
FAQ
How many strong sequence tags are enough before adding another workflow?
There is no fixed number. The better indicator is whether the current tags already define real sequence content but still fail to connect across the gap that matters for the decision.
When does top-down proteomics add more value than another digest?
Top-down proteomics becomes more useful when the unresolved question is proteoform-level, such as truncation, terminal heterogeneity, or whether bottom-up peptides belong to the same intact molecule.
Can ETD or ECD solve every PTM localization problem?
No. They can improve PTM localization when the precursor charge state and fragmentation behavior are favorable, but they do not guarantee site-level closure for every modified analyte.
Should database search still be run in a de novo project?
Usually yes. Even when the target may be novel, database-search context can help flag contaminants, detect partial homology, and eliminate implausible interpretations.
What information should a team prepare before requesting workflow advice?
Prepare the raw LC-MS/MS files, current de novo output, intact mass information if available, sample amount, purity estimate, suspected PTMs, and a short statement of the exact decision the final report needs to support.
How to order?

