





How to Identify Amino Acids and Modification Sites from a Mass Spectrum
-
A single MS1 mass usually cannot prove a full amino acid sequence; MS/MS fragment ions are needed for residue-level evidence.
-
Amino acids are inferred from mass differences between fragment ions, especially b-ion and y-ion series.
-
PTMs are detected as mass shifts, such as phosphorylation (+79.966 Da), acetylation (+42.011 Da), oxidation (+15.995 Da), or glycosylation-specific masses.
-
Modification site localization requires fragments that bracket the modified residue.
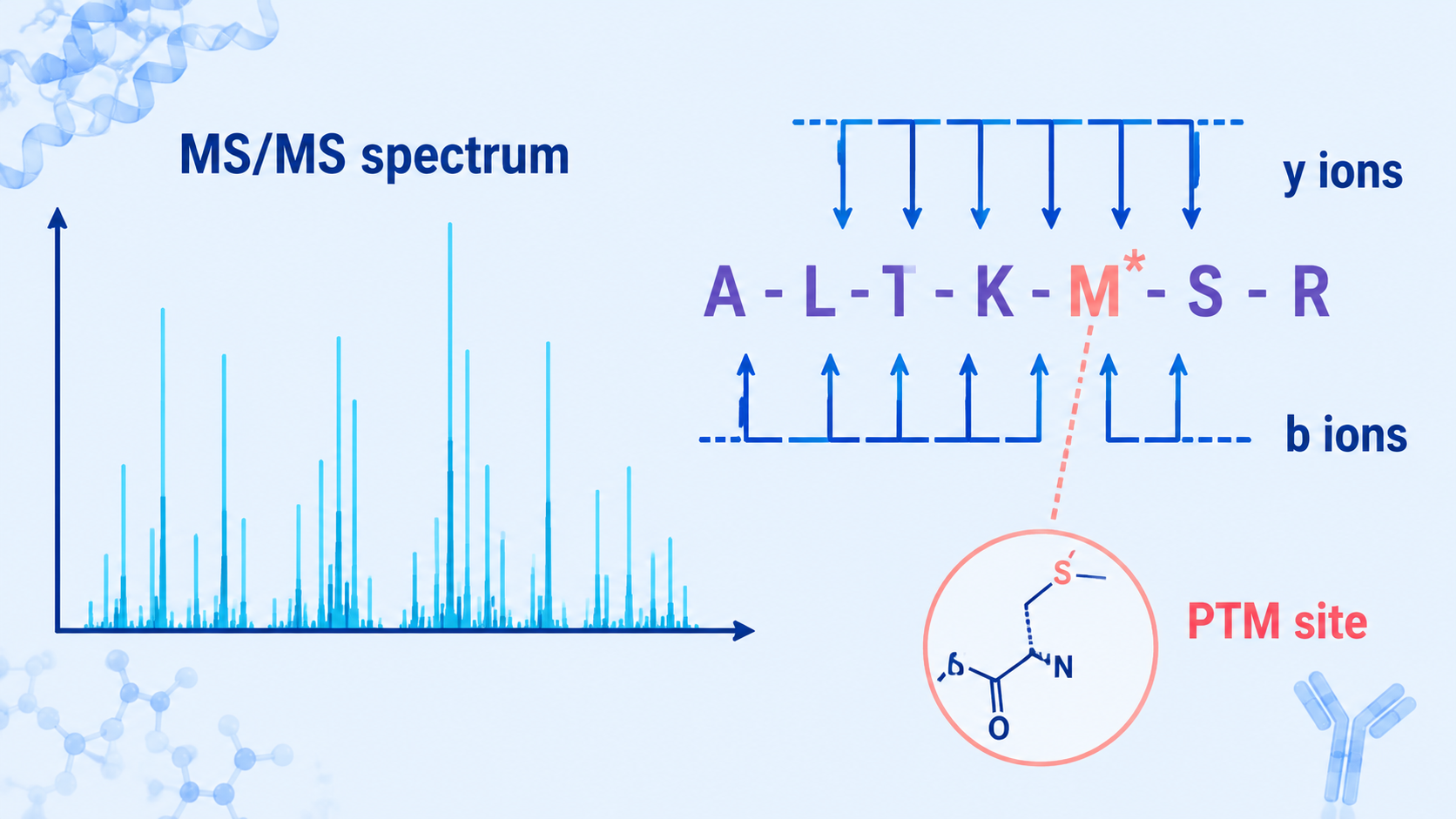
To identify which amino acids are present from a mass spectrum, analysts examine precursor mass, fragment ion series, and mass differences between adjacent MS/MS peaks. To determine a modification site, they look for a diagnostic mass shift on specific fragment ions, then compare candidate site assignments with localization scores, neutral losses, sequence context, and database or de novo evidence.
Key Takeaways
What Information Does a Peptide Mass Spectrum Provide?
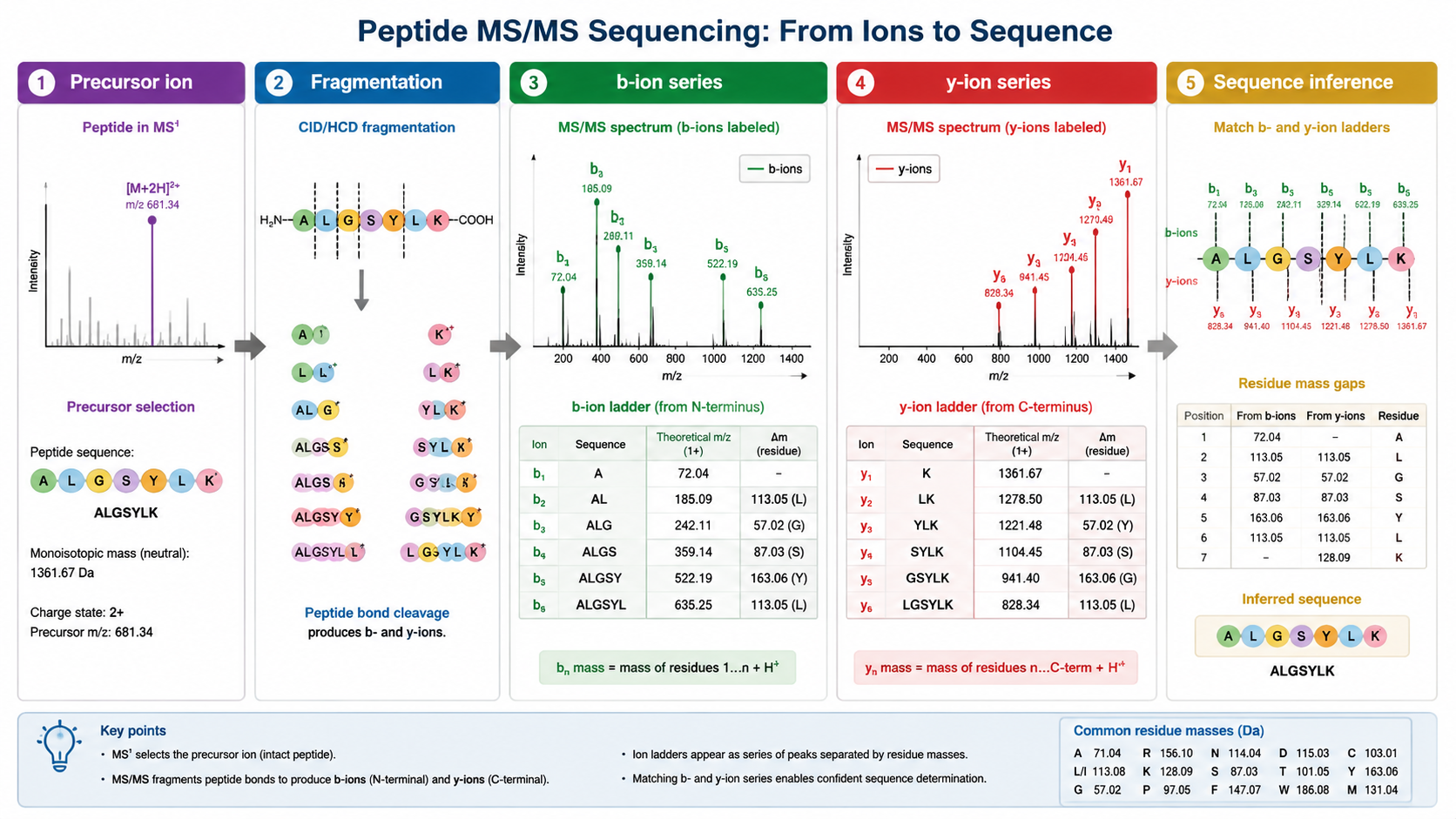
An MS1 spectrum measures precursor ions and their mass-to-charge ratios. An MS/MS spectrum fragments a selected precursor and records product ions. In peptide sequencing, the most useful ions often form b-ion and y-ion ladders. The mass difference between neighboring ions can correspond to an amino acid residue.
Related Services
Amino Acid Profile Analysis Service
Mass Spectrometry-Based Protein Sequencing Service
Mass Spectrometry-Based Protein Modification Sites Analysis Service
Step 1: Check Precursor Mass and Charge
The precursor m/z and charge state define the neutral peptide mass. This mass is compared with theoretical peptide masses from a protein database or with possible de novo sequence candidates. Accurate mass narrows the search space, but many peptide sequences can share similar masses.
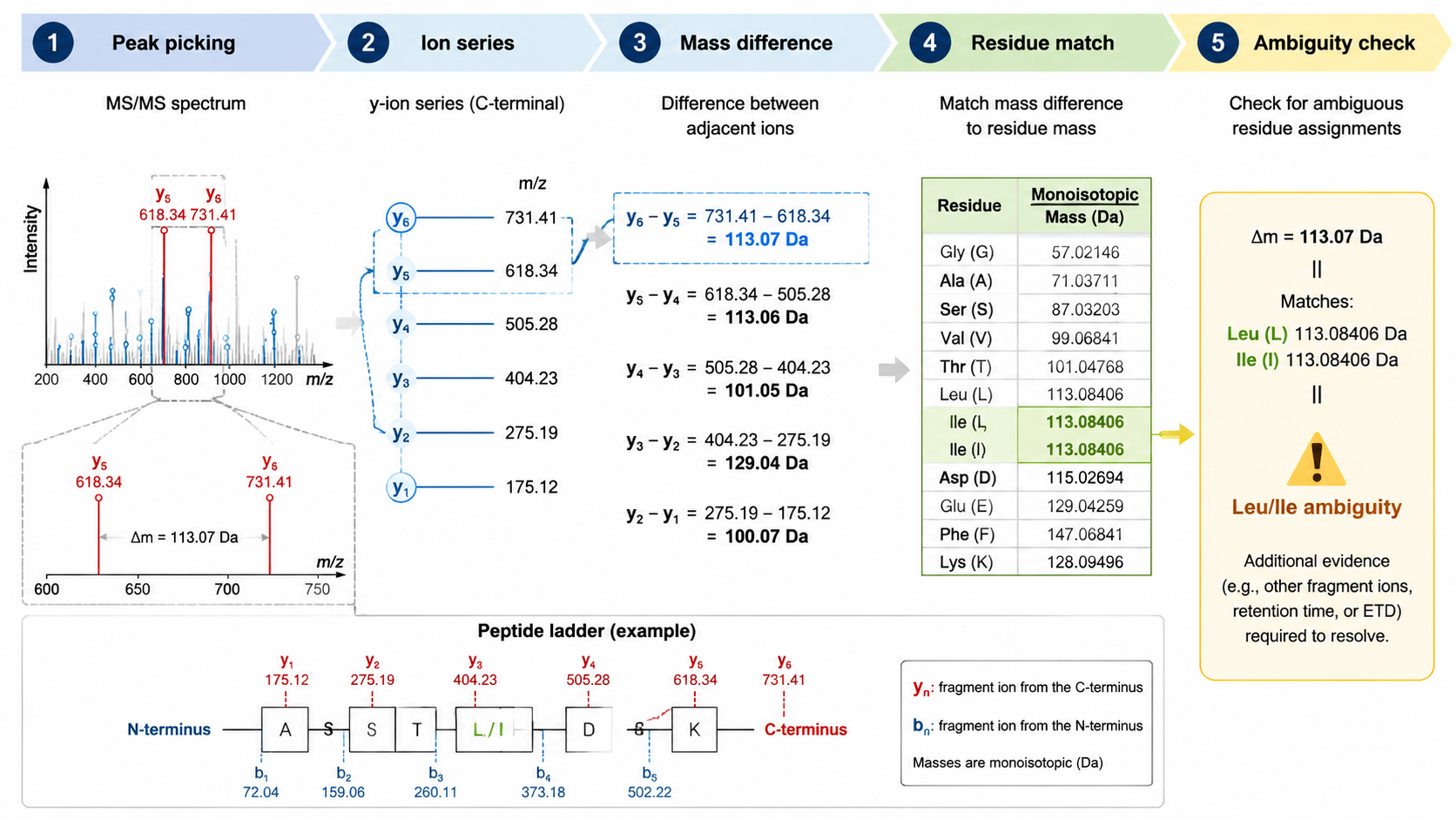
Step 2: Read Fragment Ion Mass Differences
In MS/MS spectra, adjacent b ions or adjacent y ions differ by residue masses. For example, a mass difference close to 147.068 Da suggests phenylalanine, while 129.043 Da suggests glutamic acid. Some residues are isobaric or nearly isobaric: leucine and isoleucine have identical residue masses, so MS/MS alone usually cannot distinguish them without extra evidence.
Step 3: Detect Modification Mass Shifts
A modification appears as an unexpected mass shift on the precursor, fragment ions, or both. The shift should match a plausible PTM or chemical change. Common examples include oxidation of methionine, phosphorylation of serine/threonine/tyrosine, acetylation at lysine or the protein N-terminus, and glycosylation on asparagine or serine/threonine.
Neutral losses can help interpretation. Phosphopeptides often show loss of phosphoric acid under some fragmentation conditions, while glycopeptides may show oxonium ions. These signals support the presence of a modification but do not always prove the exact site.
Step 4: Localize the Modification Site
To localize a modification, the spectrum must contain fragment ions that separate candidate residues. If two possible sites produce the same fragment pattern, the site remains ambiguous. Site localization tools compare alternative modified peptide forms and calculate confidence scores based on site-determining ions.

Practical Interpretation Table
| Evidence | What Does It Support? | What It Cannot Prove Alone? | Recommended Check |
|---|---|---|---|
| Accurate precursor mass | Candidate peptide composition | Full sequence | Verify MS/MS fragments |
| b/y ion ladder | Residue order | All isobaric distinctions | Check sequence coverage |
| Mass shift | Possible modification type | Exact site | Look for site-determining ions |
| Neutral loss | Modification class support | Exact residue assignment | Combine with fragment ions |
FAQ
1. Can you identify amino acids from only an MS1 mass spectrum?
Only partially. MS1 mass can suggest peptide composition or candidate sequences, but MS/MS fragmentation is usually required to determine amino acid order.
2. How do mass differences identify amino acids?
Adjacent fragment ions in a b-ion or y-ion series differ by the mass of one amino acid residue. Matching those differences to a residue mass table helps infer the peptide sequence.
3. How is a modification detected in MS/MS?
A modification is detected as a mass shift on the precursor and on fragment ions. Diagnostic ions or neutral losses can provide additional evidence for certain modification classes.
4. What makes a modification site confident?
A confident site assignment requires fragment ions that distinguish one candidate residue from another, plus a strong localization score and consistent spectral evidence.
Conclusion
Mass spectra identify amino acids and modification sites through patterns, not isolated peaks. Precursor mass narrows candidates, fragment ion ladders reveal residue order, and site-determining ions localize modifications. For high-value PTM sites, the safest interpretation combines automated scoring with expert spectrum review and targeted validation.
How to order?

