





Mass Spec De Novo Sequencing When Database Search Fails: A Workflow for Unknown Peptide Identification
- Confirm monoisotopic mass against the isotope envelope.
- Check that the assigned charge state explains the precursor spacing.
- Match the MS/MS event to the correct LC feature and retention time.
- Review the isolation window for competing precursor species.
- Look for consistent fragment-ion mass error across the proposed ladder.
- Prioritize continuous b-ion and y-ion series.
- Use evidence from both N-terminal and C-terminal directions when available.
- Keep only residue steps that remain plausible across more than one fragment series.
- Record unresolved positions explicitly instead of forcing a residue call.
When a peptide still cannot be resolved after an LC-MS/MS database search, the next move is usually not to keep widening search parameters. A better starting point is to check whether the precursor ion assignment is trustworthy, whether the tandem mass spectrometry data contains a readable fragment ion ladder, and whether the unresolved gap looks more like a missing database entry, a post-translational modification, or a mixed spectrum. If those checks hold up, mass spec de novo sequencing can convert unresolved MS/MS evidence into a defensible sequence tag, a candidate peptide sequence, or a clearly bounded partial answer.
The key decision is simple: are you looking at a database problem or a spectrum problem? If monoisotopic mass, charge state, and fragment-ion mass error remain internally consistent, de novo peptide sequencing is often justified. If spectral quality is weak, co-isolation is obvious, or terminal ion coverage is too sparse to support residue order, better LC-MS/MS data usually helps more than another round of database searching.
Where This Failure State Shows Up in Real Projects
This pattern usually appears after routine peptide identification was handled correctly. The raw data files are present, the precursor ion is visible, and the LC feature looks real, yet the database search produces no confident peptide-spectrum match, only weak hits, or several incompatible assignments. That situation is common in non-model organism work, proprietary peptide constructs, degradation impurities, clipped biologic fragments, venom peptides, host-defense peptides, and modified synthetic byproducts.
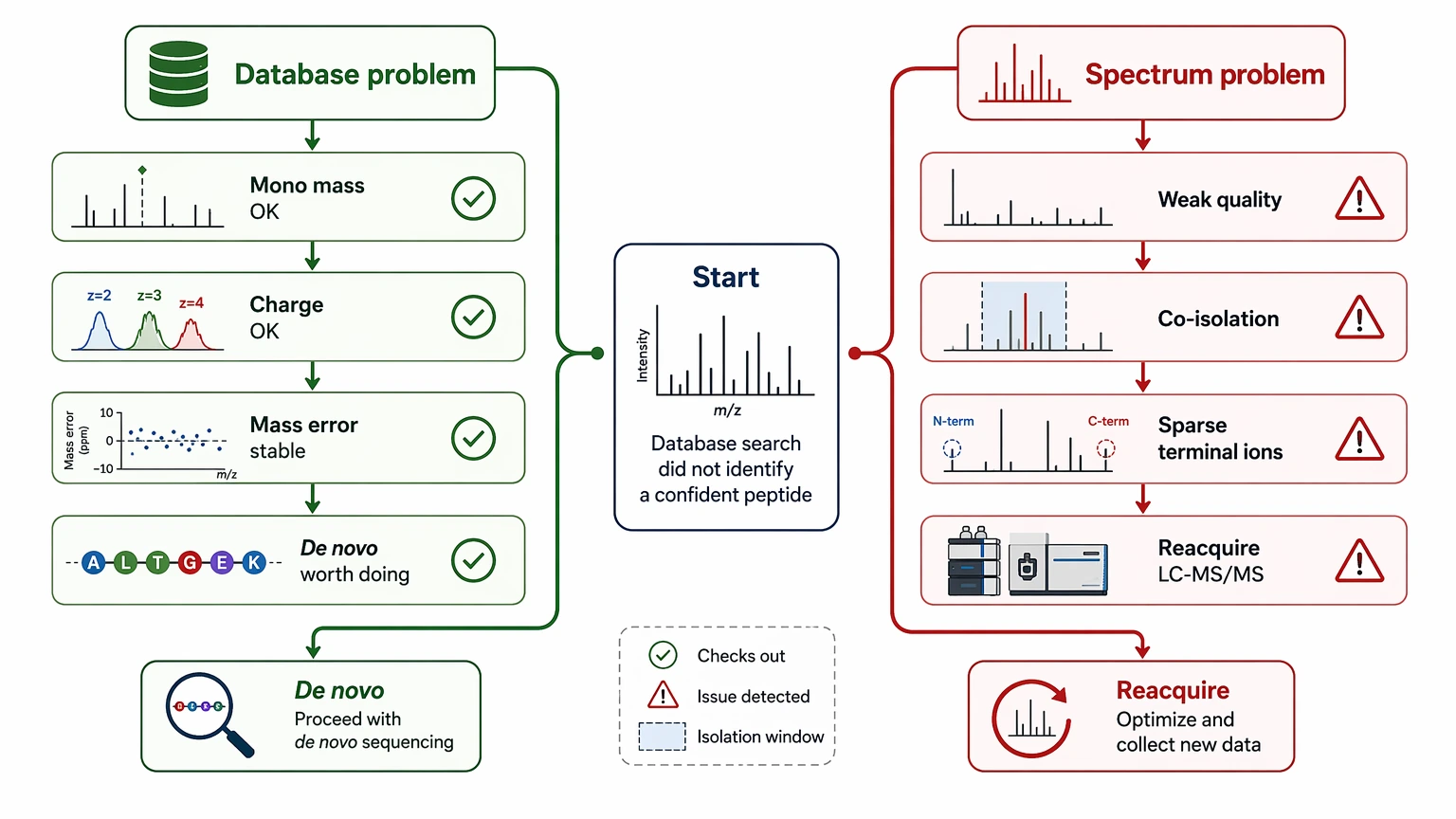
At that point, the practical question is not whether de novo peptide sequencing sounds useful. The real question is whether the available evidence can support unknown peptide identification without pushing the interpretation too far. A wrong sequence proposal can misdirect impurity follow-up, targeted MS design, or synthetic peptide confirmation. A structured workflow separates spectra that can be interpreted now from spectra that first need cleanup or reacquisition.
The Most Relevant Causes of Database Search Failure
For unresolved peptide cases, four cause categories explain most failures and point directly to the next decision.
| Cause category | Typical evidence | Best next move |
|---|---|---|
| True sequence absent from the searched database | No hit, remote partial match, or biologically implausible match | Build a sequence tag and test de novo interpretation |
| Fragmentation is incomplete or uneven | Broken b ions and y ions, long internal gaps, weak terminal coverage | Reacquire MS/MS before assigning a full sequence |
| PTM burden or an unknown mass shift breaks search assumptions | Local residue order looks plausible, but precursor mass does not reconcile cleanly | Evaluate post-translational modification and PTM localization |
| Co-isolation creates a mixed spectrum | Incompatible fragment ladders, noisy isolation window, inconsistent precursor behavior | Clean up the sample or reacquire with narrower isolation |
These categories matter more than a long generic failure list because they define whether you should keep interpreting, collect new data, or stop short of a full assignment.
A Stepwise Workflow for Unknown Peptide Identification
This is primarily a data-interpretation workflow with a project-planning checkpoint near the end. Start with evidence quality, then decide how far sequence reconstruction can realistically go.
1. Confirm that database search has actually stopped being useful
Define the failure state before switching methods. In practice, that usually means one of four conditions: no peptide-spectrum match passes the false discovery rate filter, a top-scoring hit has poor fragment support, several low-confidence assignments disagree with each other, or a nominal hit conflicts with precursor mass, sample origin, or expected processing. If the wrong FASTA database was searched or an expected modification was omitted, a focused repeat search may still solve the problem. Once broader searches keep generating unstable answers, a larger search space adds ambiguity faster than confidence.
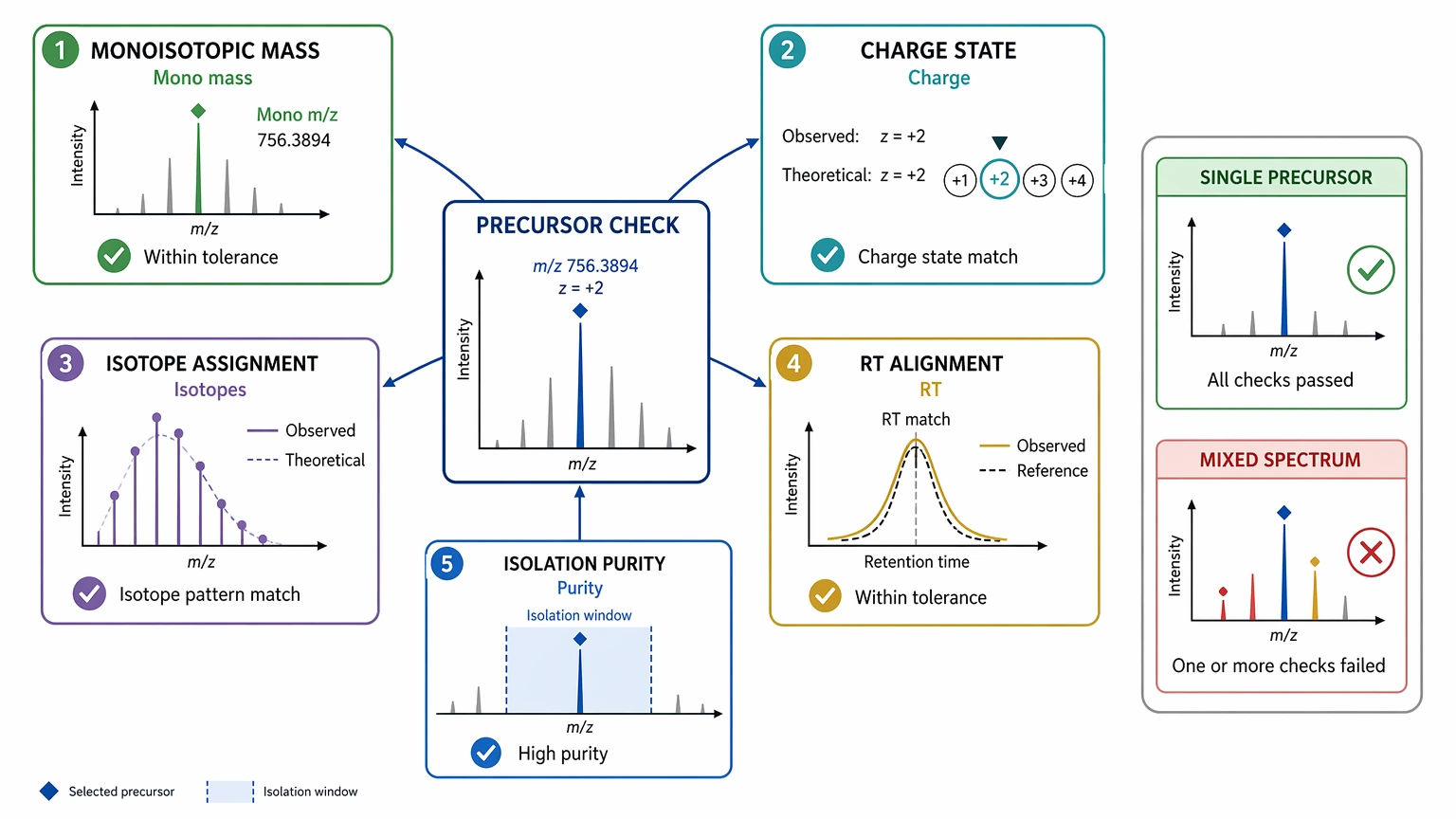
2. Verify the precursor ion before reading residues from the spectrum
De novo interpretation breaks down quickly when the precursor ion is wrong. Recheck monoisotopic mass, charge state, isotope assignment, retention-time alignment with the LC feature, and spectral purity in the isolation window. The practical question is whether the spectrum mostly comes from one precursor species. If not, sequence reconstruction should stop there because co-isolation can create a mixed spectrum that resembles a partial b-ion or y-ion ladder.
Once these checks are internally consistent, residue-level interpretation becomes much safer.
3. Extract the longest high-confidence sequence tag
Once the precursor assignment looks credible, start with the clearest fragment ion ladder instead of trying to explain every visible peak. In many unknown peptide identification projects, the most useful first deliverable is a sequence tag supported by contiguous b ions, contiguous y ions, or both. That local evidence is often more defensible than an early full-sequence guess.
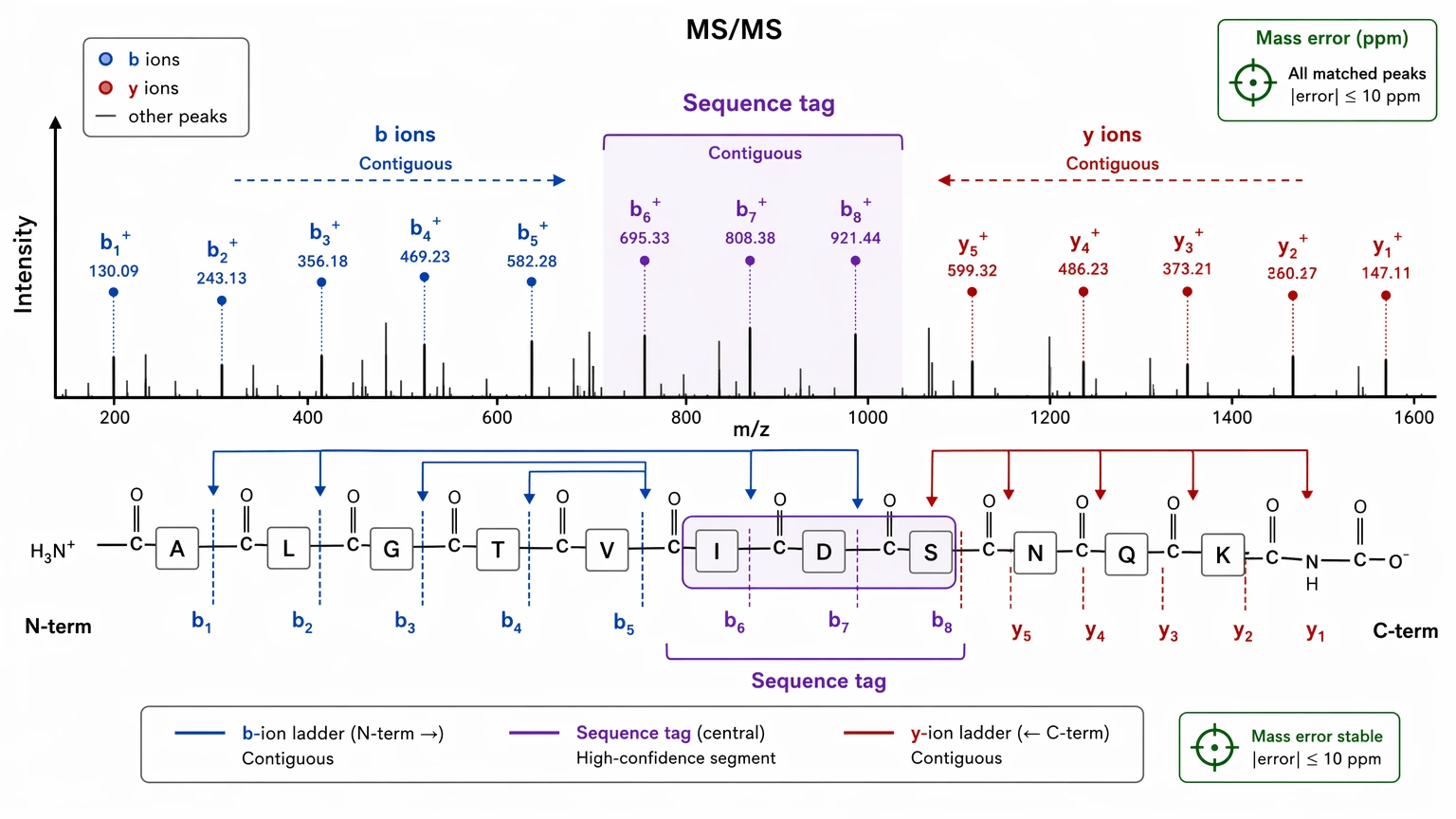
A sequence tag does not need to cover the whole peptide to be useful, but it does need to survive comparison against alternative residue paths. One boundary should remain explicit in the report: routine LC-MS/MS often cannot distinguish leucine from isoleucine because they are isobaric.
4. Reconcile the sequence tag with precursor mass and possible modifications
After extracting a sequence tag, compare the tag-derived mass with the precursor-derived monoisotopic mass. If the difference is small and localizable, the missing mass may reflect a post-translational modification, terminal chemistry change, or truncation event. If the gap is large or inconsistent across the ladder, the tag itself may need to be reconsidered. A useful classification is whether the mismatch behaves like a localized mass shift, a terminal shift, unresolved extra mass with weak PTM localization evidence, or a global mismatch large enough to challenge the proposed residue path.
This step prevents a common de novo sequencing error: accepting a locally plausible tag that does not fit the precursor ion as a whole. Final reporting should distinguish a full candidate sequence with limited PTM localization from a partial sequence tag plus unresolved modification burden.
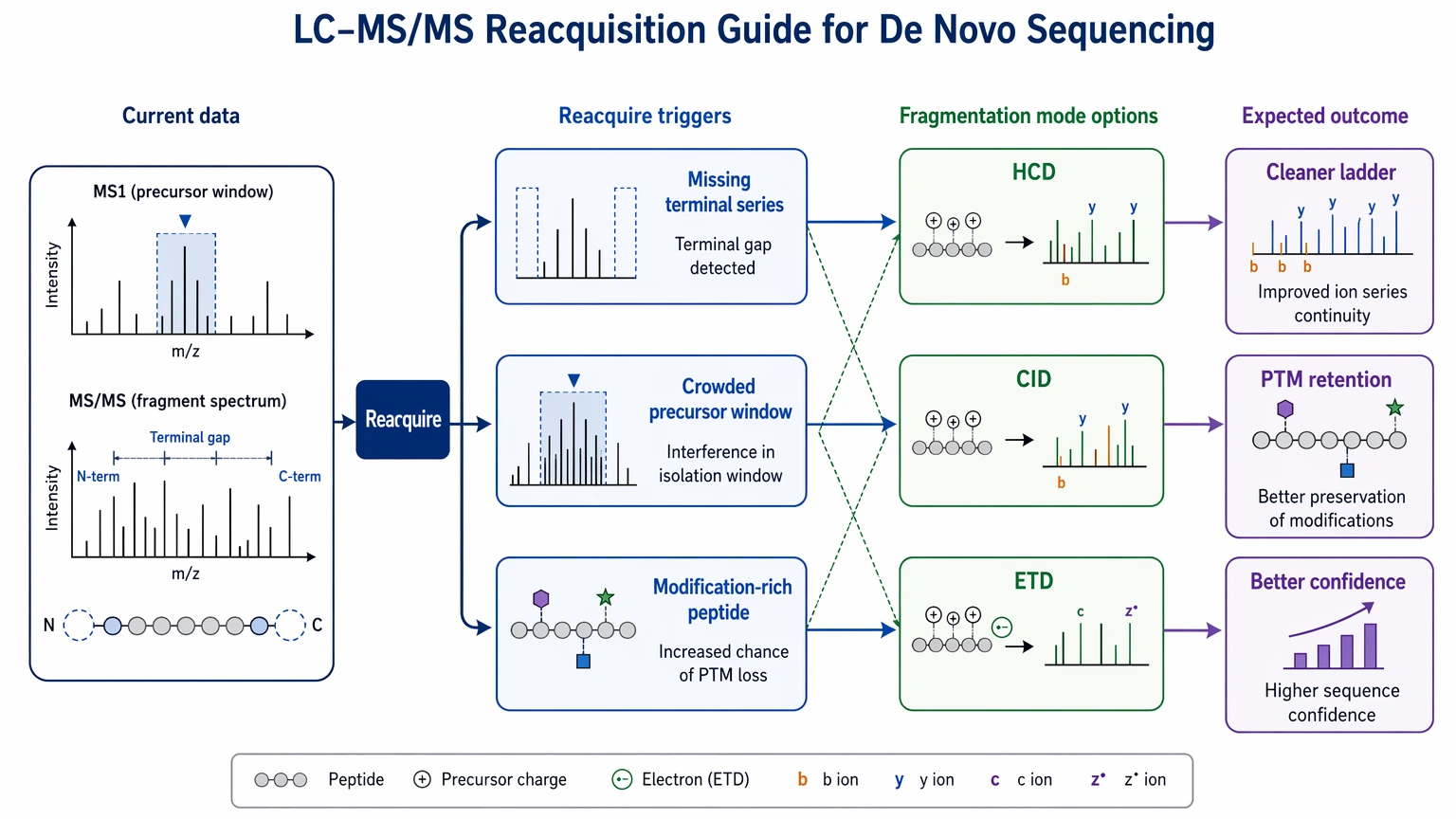
5. Decide whether more acquisition is worth more than more analysis
Before assigning a final result category, decide whether the current data are already near their interpretive limit. New acquisition usually helps when one terminal series is almost absent, the precursor window is crowded, or the peptide appears rich in modifications.
Common triggers for reacquisition include HCD data with large sequence gaps, charge states that suppress informative backbone fragmentation, clear co-isolation risk, and suspected labile modifications that fragment poorly under one mode. This is where complementary fragmentation can change the outcome. ETD may preserve labile post-translational modification information better for some multiply charged species, while HCD or CID may produce cleaner small-peptide ladders in other cases. If your team is deciding whether the current raw data files and sample amount are enough for outside review, submit your requirements and evaluate your project with MtoZ Biolabs against the likely sequence-confidence range and the need for additional acquisition.
6. Report the result in the right confidence category
Unknown peptide identification should end with an evidence category, not just a sequence string. That category guides downstream risk and determines whether targeted confirmation, more acquisition, or a stop decision is the sound next step.
| Output category | What supports it | Typical downstream use |
|---|---|---|
| Full sequence proposal | Strong terminal evidence, coherent fragment ion ladder, consistent precursor mass fit | Targeted confirmation and candidate assignment |
| Partial sequence tag | Reliable local ladder with incomplete global coverage | Family-level comparison or targeted reacquisition |
| Modification-aware candidate set | Sequence framework present, but PTM localization remains limited | Impurity or variant investigation |
| Insufficient evidence | Mixed spectrum, poor spectral quality, or irreconcilable mass pattern | Cleanup, reacquisition, or stop decision |
A strong full-sequence proposal may justify synthetic peptide confirmation. A mixed spectrum that supports only a short sequence tag usually does not.
Validation Planning After a De Novo Sequence Proposal
A de novo result becomes more useful when the validation plan matches the confidence level of the assignment. Common options include synthetic peptide confirmation for near-complete candidate sequences, targeted MS follow-up to reproduce key fragment ions and precursor behavior, intact mass cross-checks to test overall mass consistency, protease-based confirmation logic when cleavage behavior can distinguish candidates, and Edman sequencing in select cases when N-terminal evidence is decisive.
Validation matters most in biologics characterization, impurity studies, and novel peptide discovery programs where a sequence proposal may influence later development decisions.
Practical Boundaries That Should Stay Explicit
Mass spec de novo sequencing can recover useful sequence information after database search failure, but the output still depends on what the spectrum actually supports. Routine tandem mass spectrometry may leave leucine/isoleucine ambiguity unresolved. PTM localization should not be claimed from precursor mass alone. Mixed spectra can support more than one plausible residue path. In some projects, the most defensible answer is a partial sequence tag or a candidate family rather than one final sequence.
A realistic workflow also has to account for sample constraints. Repeat acquisition, orthogonal fragmentation, and validation studies often require more material than the first discovery run used. If the unknown belongs to a larger protein context rather than a stand-alone peptide, bottom-up and top-down strategy fit should be reconsidered before more residue-level interpretation is attempted.
Service Routes to Consider
If the main decision is whether the current data can support de novo interpretation or should move to reacquisition and confirmation, these service paths fit that checkpoint well.
Conclusion
When database search fails, the most productive next step is to test whether the precursor ion, fragment ion ladder, and global mass balance support de novo interpretation in the first place. The practical workflow is to verify precursor assignment, extract a defensible sequence tag from b ions and y ions, reconcile that tag against monoisotopic mass and possible post-translational modification burden, decide whether HCD, CID, or ETD reacquisition is needed, and then report the result as a full sequence proposal, partial sequence tag, modification-aware candidate set, or insufficient evidence. That framework fits impurity characterization, clipped peptide analysis, non-model organism discovery, and other unknown peptide identification projects where routine peptide-spectrum match logic has stopped helping. If your team needs to judge whether the raw data files, sample purity, and validation goals are sufficient for a de novo study, contact us to discuss the case with MtoZ Biolabs, evaluate your project, and plan the most appropriate orthogonal validation path before the result is used in a higher-stakes decision.
FAQ
How do I tell whether the database itself was the problem?
Check biological fit as well as score. If the best peptide-spectrum match comes from an unrelated species, an implausible precursor mass offset, or a sequence that conflicts with known construct design, database coverage may be the real issue.
What sample information is most useful before starting de novo peptide sequencing?
Raw data files, the precursor list, sample source, purification status, prior database search settings, and any suspicion of truncation or post-translational modification are the most helpful inputs.
When is a partial sequence tag already enough to move a project forward?
A partial tag can be enough to group an impurity, narrow a peptide family, design targeted follow-up MS, or decide whether synthesis of a small candidate set is justified.
Should I merge replicate spectra before manual interpretation?
Replicates help only if they confirm the same precursor ion, charge state, and fragment-ion pattern. They can mislead if they combine slightly different species or different co-isolation backgrounds.
Is de novo sequencing still worthwhile for a peptide mixture?
Sometimes, but only if one precursor can be isolated cleanly enough to avoid a persistent mixed spectrum. If multiple coeluting species keep entering the same isolation window, cleanup or narrower targeting usually improves confidence more than deeper interpretation.
Related Services
Main Service |
Supporting Service |
Validation Service |
Alternative Service |
How to order?

