





Hybridoma Monoclonal Antibody Sequencing for Legacy Clones: How to Recover VH/VL Sequences When Cell Records Are Incomplete
- Is there one productive VH sequence and one productive VL sequence, or several candidates?
- Is an aberrant light chain present?
- Do the candidate sequences fit the known isotype and expected antibody architecture?
- Is sequence coverage complete enough for cloning and recombinant re-expression?
- Preliminary sequence recovery: plausible immunoglobulin sequence information, but incomplete for operational use
- Documentation-grade output: sufficiently annotated sequence evidence for archive rebuilding or internal review
- Re-expression-ready output: paired VH/VL with adequate completeness and validation support for cloning
Recovering antibody sequence information from a legacy hybridoma clone is not just a matter of finding an immunoglobulin-like read. What matters is showing that the final paired VH/VL assignment is the correct one for downstream use. In most cases, the most defensible path starts with the strongest remaining biological evidence, uses transcript-based sequencing when cryopreserved hybridoma cells or usable RNA are still available, and adds orthogonal validation before recombinant re-expression, transfer, or documentation.
That distinction matters when records are incomplete. A partial heavy chain variable region, a single CDR call, or a short list of possible light chain candidates can all look promising in a report, but none of them automatically means the clone is ready for re-expression. For teams working with older murine assets, the real question is whether the recovered VH sequence and VL sequence are complete, productive, and credibly paired to the antibody that still matters to the program.
Where Sequence Recovery Becomes a Project Risk
This problem often shows up late in an antibody program. A team may want to reformat an older clone into recombinant IgG, transfer the asset across sites, rebuild an archive, or support internal IP review. Then the record trail stops being reliable: freezer labels are ambiguous, notebook entries are incomplete, the stored vial may not match the historical data package, or only culture supernatant or a purified monoclonal antibody lot is left.
At that point, hybridoma monoclonal antibody sequencing turns into a risk-management exercise rather than a routine sequencing request. A sequencing dataset may show one dominant VH sequence, several VL candidates, nonproductive transcripts, or an aberrant light chain signal. If those findings are pushed forward too quickly, the wrong chain assignment can move into cloning and create expensive rework later for R&D, tech transfer, and documentation teams.
The Failure Modes That Matter Most
1. Uncertain clone provenance
A legacy hybridoma clone is only useful if the current material really represents the historical antibody asset. Mislabeling, undocumented recloning, passage drift, or contamination can weaken that connection. Even a technically clean sequence result has limited value if the sample itself is not clearly tied to the intended clone.
2. Starting material constrains the recovery path
Cryopreserved hybridoma cells, RNA, culture supernatant, and purified monoclonal antibody do not support the same level of inference. Transcript-based sequencing can directly inform heavy chain variable region and light chain variable region recovery. Protein-only material can still support sequence recovery, but usually with less direct evidence for native chain pairing.
3. Background transcripts create false confidence
Hybridomas can contain productive immunoglobulin transcripts alongside nonproductive species or aberrant light chain background. That means a next-generation sequencing (NGS) dataset may return several plausible candidates instead of one obvious answer. In legacy projects, interpretation often matters as much as data generation.
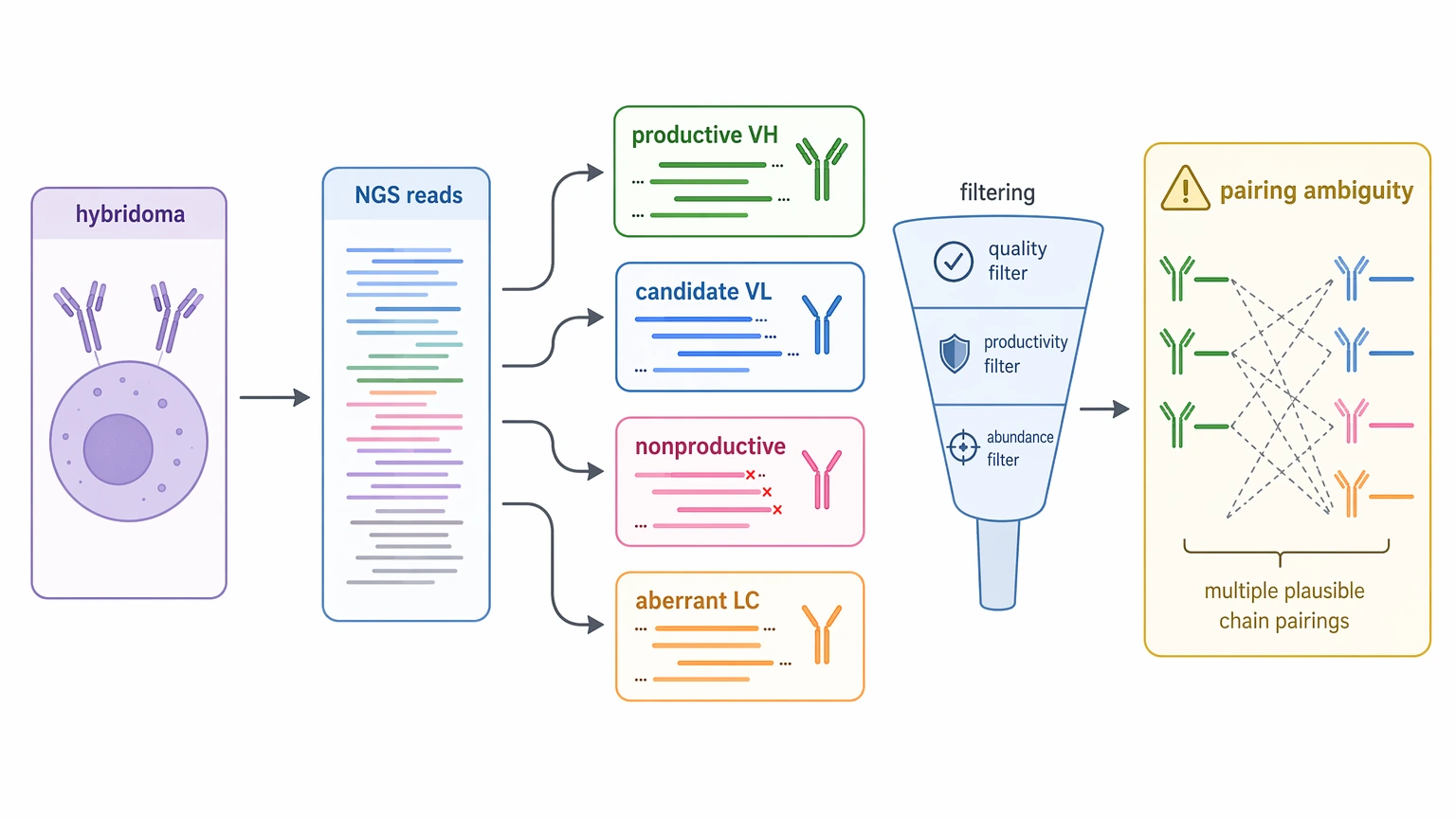
4. Partial reads are mistaken for operational readiness
A CDR-focused result can help with early identity checks, but recombinant re-expression usually requires full variable-region coverage with continuous framework region definition and no unresolved residues at critical positions.
5. Validation comes too late
Teams sometimes treat sequence recovery as finished before checking isotype confirmation, peptide mapping, or binding concordance. That shortcut tends to surface later, when recombinant material does not match the original antibody behavior.
A Project-Planning Workflow for Hybridoma Monoclonal Antibody Sequencing
Step 1: Define what the sequence must support
Start with the endpoint, not the assay. A documentation-grade deliverable is not the same as a re-expression-ready one. If the goal is recombinant re-expression, the usual target is paired VH/VL with productive coding sequence, strong framework region coverage, and enough confidence in chain pairing to build vectors without guessing at missing residues.
This first decision prevents the team from accepting an answer that looks good but is still incomplete. A plausible antibody-like sequence may be enough for archive rebuilding, while reformatting or transfer may call for a higher level of evidence.
Step 2: Inventory every surviving material source
Collect all remaining inputs before choosing a method: cryopreserved hybridoma cells, cell pellets, total RNA, cDNA, culture supernatant, purified monoclonal antibody, isotype notes, historical binding data, and any earlier sequencing traces or gel records. Then rank them by how directly they support sequence recovery and chain assignment.
| Starting material | Best-fit method | Main strength | Main limitation |
|---|---|---|---|
| Cryopreserved hybridoma cells | transcript-based sequencing, RACE-PCR, NGS | strongest route to paired VH/VL | viability or transcript quality may be reduced |
| Cell pellet or RNA | RACE-PCR, targeted amplification, NGS | good variable-region access | degradation can limit completeness |
| Culture supernatant | limited protein-based support | confirms expressed antibody presence | weak direct chain pairing evidence |
| Purified monoclonal antibody | de novo antibody sequencing, LC-MS/MS, peptide mapping | usable when cells or RNA are unavailable | isobaric ambiguity and less transcript context |
| Mixed evidence package | combined transcript + protein workflow | supports cross-checking | requires more interpretation |
Step 3: Choose the primary route from the strongest evidence
If cryopreserved hybridoma cells or workable RNA are available, transcript-based sequencing is usually the preferred starting point. RACE-PCR can help capture variable-region boundaries, and NGS can show whether multiple heavy or light chain candidates are present. That broader view is useful because it exposes ambiguity instead of hiding it.
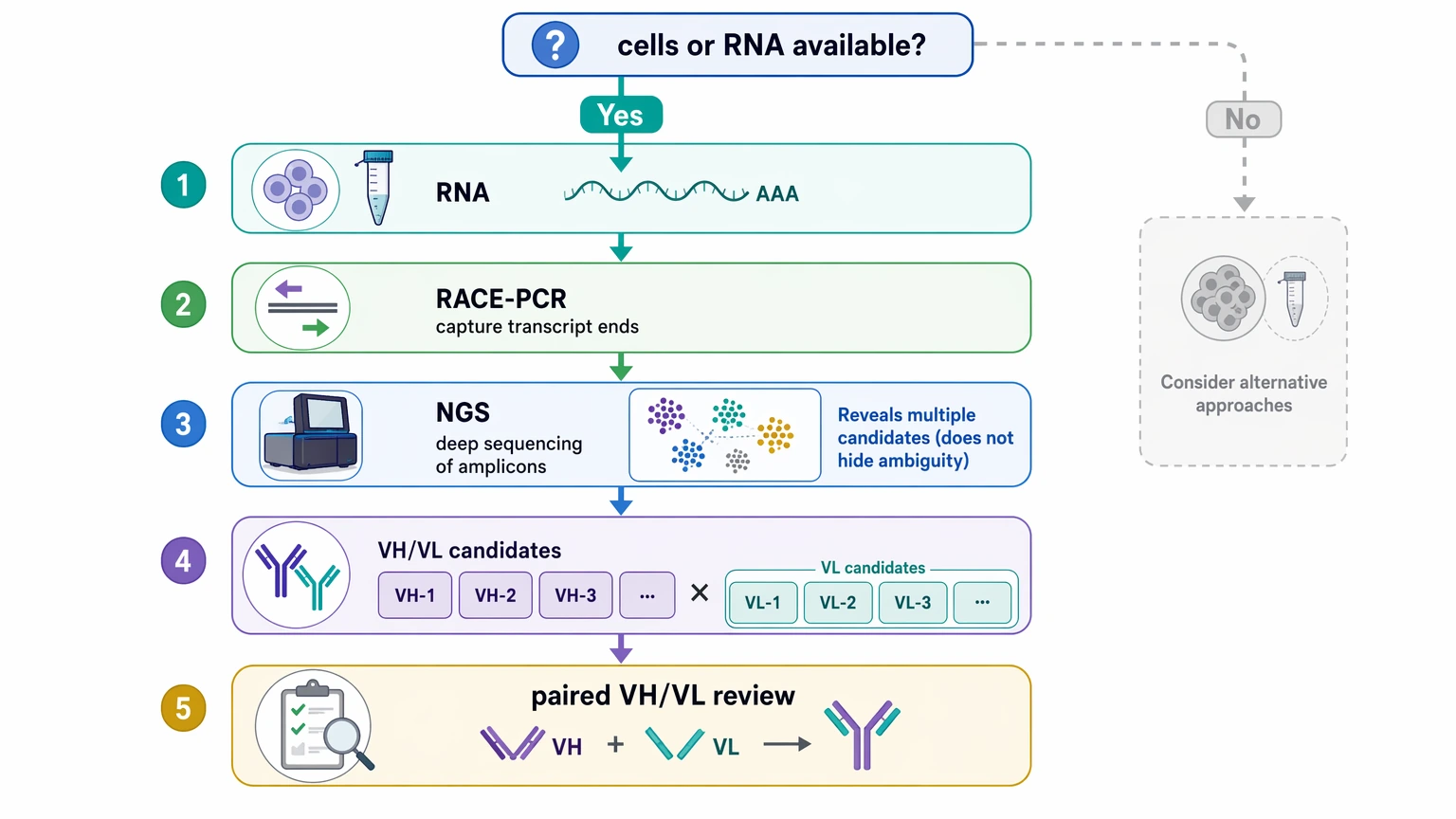
If cellular material is gone, de novo antibody sequencing by LC-MS/MS from a purified monoclonal antibody becomes more important. This route can recover much of the heavy chain variable region and light chain variable region, but teams should read the final report carefully for residue uncertainty, incomplete coverage, and limits on chain pairing confidence.
When sample history is messy, a combined workflow is often easier to defend than repeating one method in isolation. Historical isotype notes, partial transcript evidence, and peptide mapping may not solve the whole problem on their own, but together they can narrow the correct paired VH/VL choice. If your group is at that decision point, submit your requirements to evaluate your project scope before using irreplaceable material.
Step 4: Screen for wrong chain assignment before cloning
Before a recovered sequence moves downstream, review four checkpoints:
This is where many legacy projects move from 鈥渟equence obtained鈥?to 鈥渟equence not yet ready.鈥?Strong chain assignment usually depends on converging evidence: productive reading frame, transcript abundance, framework region continuity, isotype consistency, and peptide-level support when protein data are available.
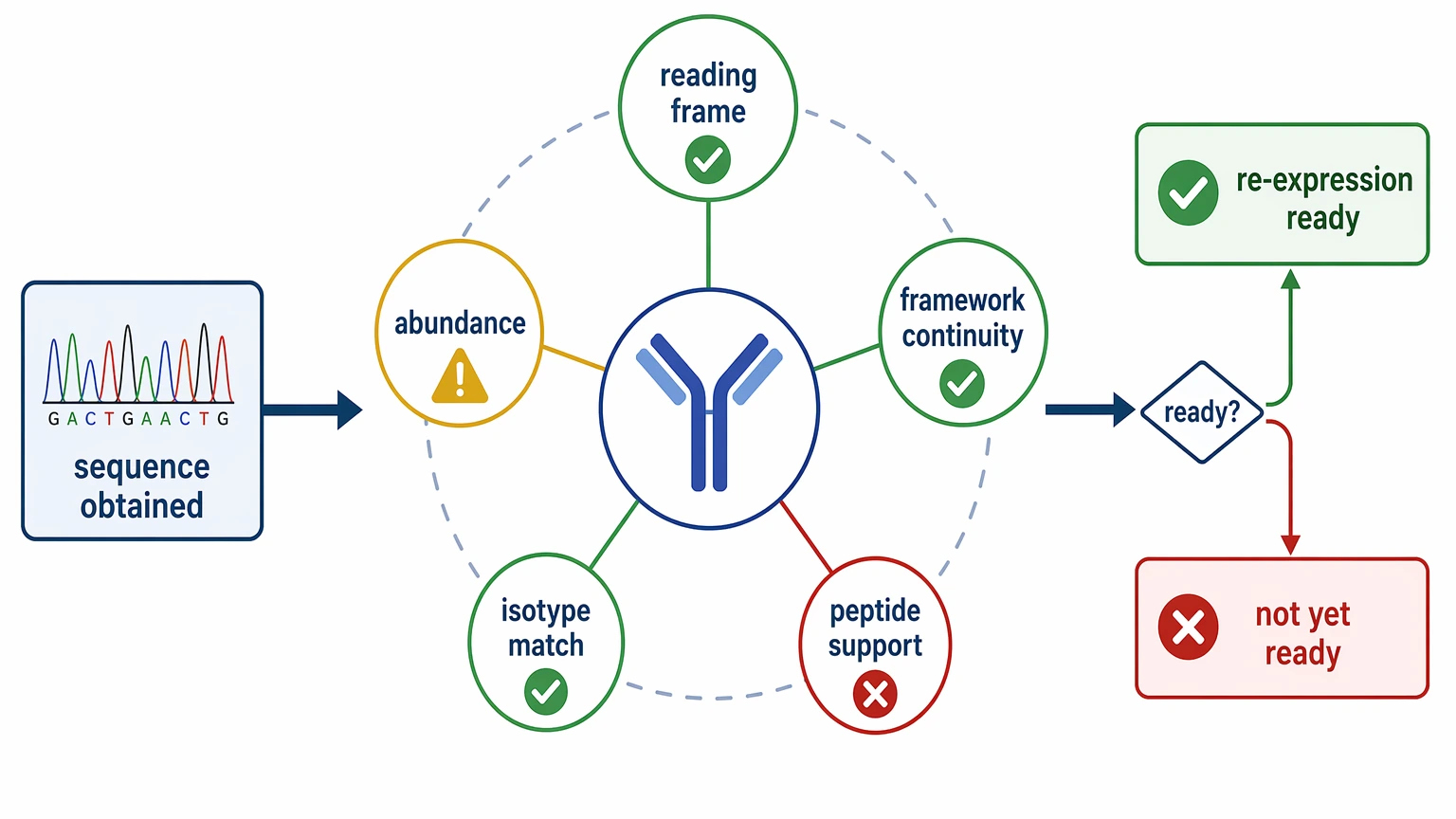
For inherited clone portfolios or transfer packages, MtoZ Biolabs can help evaluate whether the available evidence supports sequence recovery only or whether it is mature enough for recombinant re-expression planning.
Step 5: Build validation into the plan before operational use
Sequence recovery should end with a validation design, not just a sequence table. The most practical confirmation step is recombinant re-expression followed by comparison with the original antibody. Binding concordance is especially informative because it tests whether the selected heavy and light chain pair reproduces the expected binding behavior.
A focused validation package may include:
| Validation item | What it supports | When it matters most |
|---|---|---|
| Isotype confirmation | consistency with historical class information | records are incomplete or inconsistent |
| Peptide mapping | peptide-level support for recovered sequence | protein-based recovery contributed heavily |
| Binding concordance | functional agreement between original and recombinant antibody | re-expression or transfer is planned |
| Sequence completeness review | readiness for vector construction | unresolved residues remain a concern |
How to Judge Whether the Deliverable Is Actually Usable
A useful report should do more than list candidate sequences. It should lay out the chain assignment logic, identify where ambiguity still sits, and state whether the output contains productive paired VH/VL, full framework region coverage, and any unresolved residues. If several candidate light chains remain, the report should say that plainly rather than forcing a single answer without support.
Teams should also separate three deliverable levels:
That distinction keeps programs from moving too fast. A sequence can be scientifically plausible and still be too uncertain for transfer or recombinant re-expression.
Practical Execution Notes for Legacy Clones
Older material often produces mixed signals. Cryopreserved hybridoma cells may have low viability but still contain recoverable transcripts. Degraded RNA may support partial sequence recovery while leaving gaps at variable-region boundaries. Purified monoclonal antibody can preserve a rescue option when no cells remain, but it usually makes orthogonal validation more important.
Lot control matters too. If several freezer vials or antibody lots exist, document which one is being sequenced and which one will serve as the comparator for binding concordance. Otherwise, differences between lots can be mistaken for clone drift or analytical inconsistency.
Teams should also avoid forcing progress when ambiguity remains. Multiple VL candidates, mixed-clone signatures, or unresolved framework residues are signals to pause and strengthen the evidence package rather than rushing into vector construction.
Conclusion
For a legacy hybridoma clone with incomplete records, the most defensible route is to start with the strongest surviving material, recover paired VH/VL with close attention to chain assignment and completeness, and confirm the result with orthogonal validation before operational use. This approach fits archive rebuilding, recombinant reformatting, cross-site transfer, and internal documentation projects where the antibody still has value but clone provenance is no longer clean. If your team needs to sort sample options, define a sequence recovery path, or compare documentation-grade versus re-expression-ready output, contact us at MtoZ Biolabs to discuss the sample scenario and evaluate your project before committing material.
FAQ
If my hybridoma stock is nonviable, is the project still worth starting?
Often yes, if other evidence remains. Nonviable cryopreserved hybridoma cells may still yield usable RNA, and purified monoclonal antibody can support de novo antibody sequencing when transcript recovery is weak.
Do I need leader sequence recovery for recombinant re-expression?
Not always, but it can be helpful. Many re-expression projects can proceed with confident variable-region coding sequence, while leader or signal peptide information may matter more for rebuilding the original transcript context.
How should I handle a report with two plausible VL candidates?
Treat that as an unresolved chain pairing problem, not a minor annotation issue. The next step is usually additional orthogonal validation, such as peptide mapping or recombinant testing of shortlisted pairs.
Is a CDR-only result enough for transfer documentation?
Sometimes for preliminary identity tracking, but usually not for transfer-ready documentation. Most transfer packages need clearer chain-level completeness and a better record of how the final VH sequence and VL sequence were selected.
When does purified monoclonal antibody become the primary source?
It becomes the primary source when viable cells, RNA, or cDNA are unavailable or too compromised to support reliable transcript-based sequencing. Even then, teams should still use any historical isotype or binding data to strengthen interpretation.
What project details should be prepared before contacting a sequencing provider?
Prepare the sample type, number of vials or lots, known isotype, intended downstream use, any historical binding data, and whether the goal is documentation, recombinant re-expression, or transfer. That information makes workflow selection faster and reduces the risk of consuming the wrong material.
How to order?

