





Hybridoma Antibody Sequencing: How to Recover Monoclonal Antibody Sequences for Recombinant Re-Expression
- Proceed with protein-first recovery when you still have purified monoclonal antibody material with usable integrity, enough input for LC-MS/MS, and a clear reason to move toward a sequence-defined reagent.
- Proceed cautiously when the sequence evidence is likely to include unresolved isobaric residues, incomplete sequence coverage, uncertain chain assignment, or interference from post-translational modification (PTM) signals.
- Pause and seek a different rescue path first when the sample is highly degraded, mixed, contaminated, or too limited to support repeat analysis and downstream confirmation.
- usable input amount and concentration
- purity and degradation state
- likelihood of mixed antibody content
- expected interference from salts, stabilizers, or excipients
- whether repeat injections or repeat digests remain possible
- multiple digestion conditions to improve overlapping peptide evidence
- bottom-up proteomics by LC-MS/MS
- chain-aware interpretation to improve chain assignment
- intact mass / subunit analysis for consistency checks
- targeted review of peptides that span or flank CDR-containing regions
- proposed heavy chain and light chain amino acid sequences
- annotated confidence across the variable region and framework region
- explicit flags for unresolved positions, especially leucine/isoleucine ambiguity
- notes on suspected post-translational modification (PTM) effects, including oxidation, deamidation, pyroglutamate formation, clipping, or glycosylation heterogeneity
- a summary of how much of the sequence depends on true de novo sequencing versus database-assisted interpretation
- targeted peptide mapping of uncertain sites
- repeat digestion or repeat MS/MS acquisition for weak regions
- additional intact mass / subunit analysis
- limited recombinant expression of the proposed chains
- binding or comparability testing against the original antibody
When the original hybridoma is no longer a dependable source, hybridoma antibody sequencing can turn the remaining protein material into a realistic path toward recombinant re-expression. The real issue is not whether the antibody worked in the past. It is whether the material you still have can support a defensible reconstruction of the heavy chain and light chain, especially across the variable region, with uncertainties stated clearly enough to justify cloning and follow-up testing.
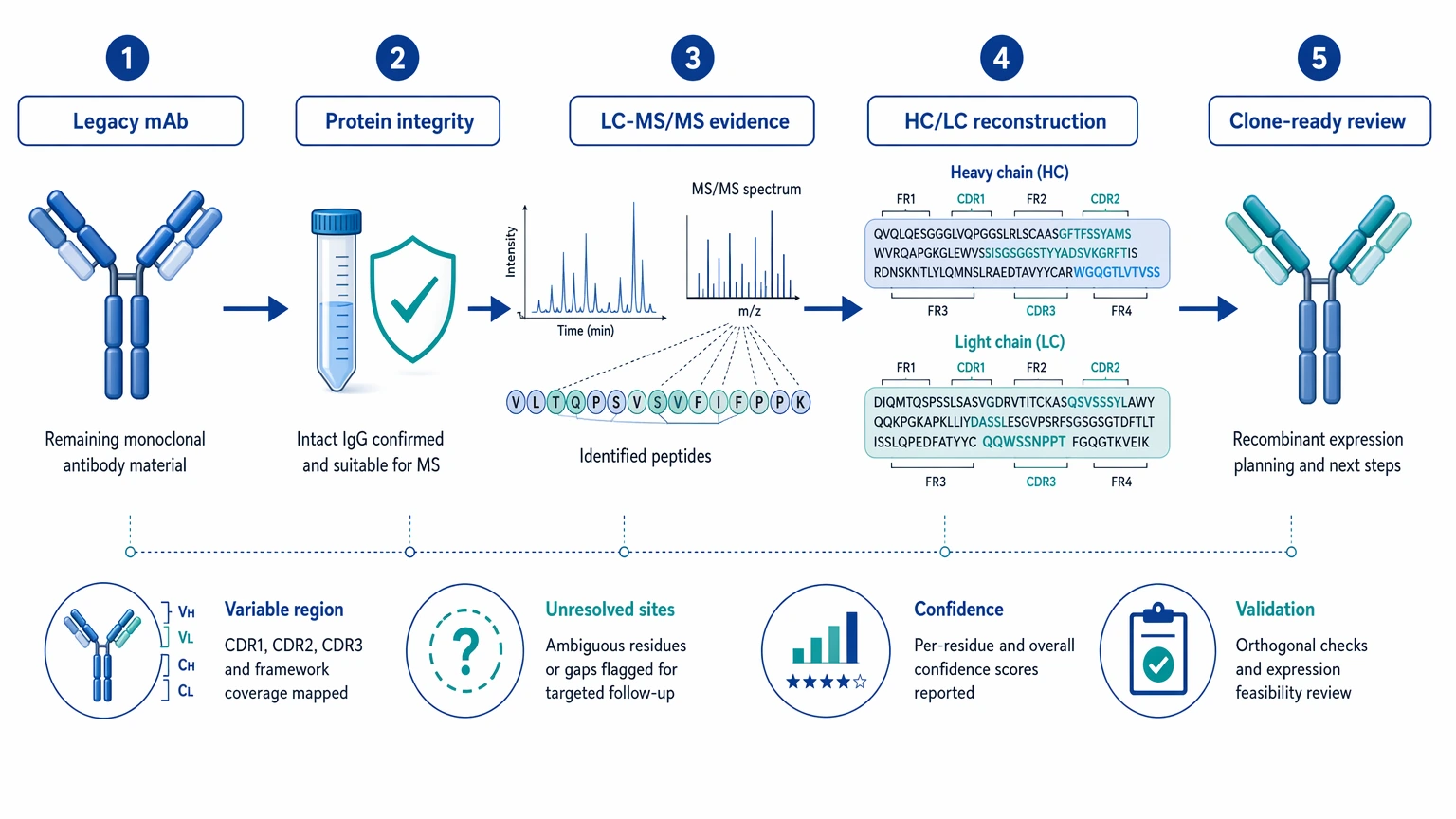
Quick decision guide
Where This Problem Usually Appears
This decision usually shows up late in a program, not at discovery kickoff. A legacy reagent may still work in binding assays, cell staining, or internal reference testing, while the original hybridoma is no longer reliable enough for routine use. Common triggers include aging cell banks, unstable subclones, incomplete transfer records, missing VH/VL documentation, or a project handoff that now requires sequence-defined materials.
At that stage, teams are not asking for broad antibody characterization. They need sequence information they can actually use to rebuild the antibody in a recombinant format. If nucleic-acid records are missing or no longer trusted, a protein-first route starts to make sense because the only remaining evidence may be in purified antibody stock, archived fractions, or older analytical material.
When a Protein-First Rescue Path Makes Sense
A protein-first strategy is most useful when the operational goal is specific: recover enough chain-level sequence information to support recombinant re-expression and a defined validation plan. That is a different task from a general identity check.
The starting material usually falls into one of the following groups:
| Sample type | Best fit | Main constraint | Practical next step |
|---|---|---|---|
| Purified intact IgG | Standard recovery planning | Buffer additives or low purity can interfere | Review concentration, purity, and formulation background |
| Reduced chains or subunits | Better support for chain-level interpretation | Extra handling can increase degradation risk | Pair with peptide mapping and subunit review |
| Fab or F(ab')2 fragments | Useful when full IgG is unavailable | Full chain context may be incomplete | Confirm which regions remain represented |
| Archived low-volume stock | Rescue projects with few alternatives | Limited repeats may restrict ambiguity resolution | Design a minimal-consumption workflow |
| Old supernatant-derived prep | Possible in some legacy cases | Host-cell protein contamination may be high | Screen enrichment and purity before full sequencing |
A simple rule helps. If the sample can generate interpretable tandem mass spectrometry data and you can still reserve material for confirmatory work after the first sequence proposal, the project may be worth moving forward. If not, recovering cleaner protein or restoring access to the hybridoma may be the faster path.
A Project-Planning Workflow for Sequence Recovery
1. Define the decision output before ordering the work
Start with the deliverable you actually need. For legacy rescue, the useful endpoint is not just a peptide list. It is a proposed full-length amino acid sequence for both chains, confidence notes by region, a record of unresolved positions, and a plan for orthogonal validation before cloning.
That changes the success criteria. Broad identification without variable-region confidence may help with characterization, but it does not automatically support rebuilding.
2. Review the sample as analytical material, not just as a historical asset
Project history matters, but sample condition usually matters more. Protein-level recovery depends on whether the antibody can generate overlapping peptides and interpretable spectra across the regions that matter. Degradation, formulation background, co-purified proteins, and low input can all weaken de novo sequencing performance.
For antibodies pulled from older inventories, the review should stay focused on a short list of factors that actually affect the decision:
3. Match the sequencing strategy to the evidence gap
For unknown or poorly documented antibodies, de novo sequencing usually matters more than database-only matching. A database search can help anchor conserved regions, but it may miss undocumented variation in the framework region and, more importantly, in the complementarity-determining region (CDR).
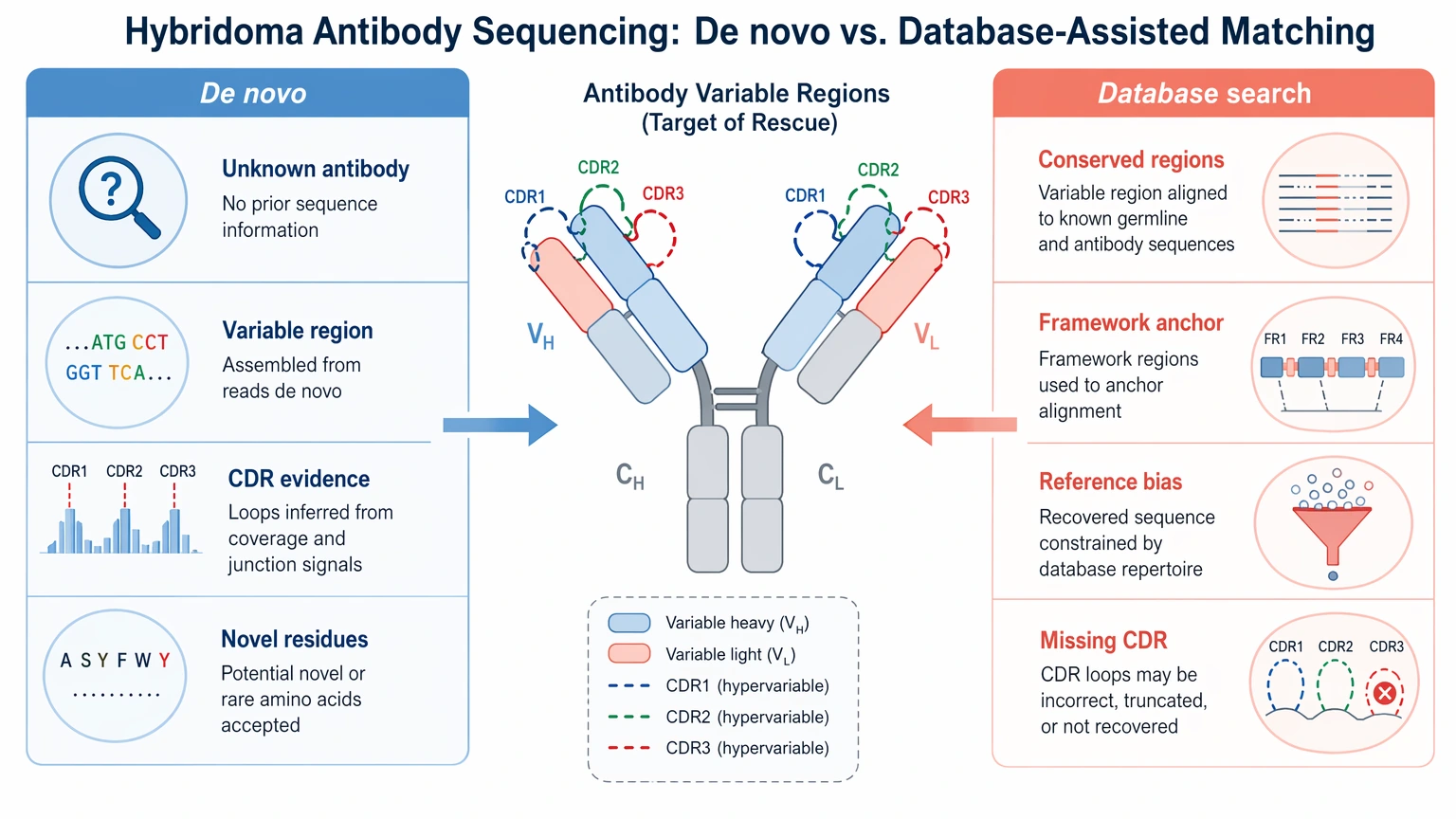
A practical recovery design often combines:
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
One limit should be explicit from the start: bottom-up MS alone may not resolve every residue with the same confidence. Leucine/isoleucine ambiguity is the standard example, and PTM-related mass shifts can complicate local interpretation further.
4. Judge the sequence package by reconstruction quality, not peptide count
A large peptide count can look reassuring while leaving the main re-expression risk unresolved. The more useful question is whether the evidence supports coherent chain reconstruction across the regions that matter for function.
| Evidence area | What it supports | Remaining boundary | Follow-up value |
|---|---|---|---|
| Broad sequence coverage across both chains | Overall reconstruction | Coverage may still be uneven in CDR-rich segments | Helps prioritize missing regions |
| Overlapping peptides in the framework region | Local sequence confidence | Strong framework evidence does not guarantee CDR certainty | Stabilizes nearby calls |
| CDR-containing peptide evidence | Re-expression relevance | Ambiguous residues may remain | Guides cloning risk review |
| Intact mass / subunit analysis | Chain-level mass consistency | Cannot assign every residue by itself | Supports chain completeness checks |
| PTM review | Distinguishes modification from sequence | Some mass shifts remain interpretation-sensitive | Helps avoid false residue calls |
A strong deliverable does not pretend to be more certain than the data allow. It shows what is well supported, what remains provisional, and which positions need another look before the sequence is treated as clone-ready.
5. Build confirmation into the plan before cloning starts
Protein-derived recovery is most useful when it feeds into a staged validation plan. If your team is deciding whether the remaining antibody stock is suitable for this route, you can submit your requirements and sample context to MtoZ Biolabs for a project-level review that aligns the sequencing workflow with the re-expression goal rather than a generic characterization request.
That planning step matters because sequence recovery and biological confirmation are related, but they are not the same milestone.
Expected Results and Validation Methods
The immediate output should be a chain-resolved sequence proposal, not just a search report. In practical terms, the first deliverable should include:

Follow-up confirmation comes next and should be treated as a separate evidence layer. Depending on the project, that may include:
That distinction matters. The first deliverable tells you whether cloning is reasonable to attempt. The follow-up work tells you whether the proposed sequence behaves as expected after re-expression.
Key Cautions and Practical Limits
Protein-first recovery can be very useful, but the boundaries need to stay visible from start to finish.
Sample quality or amount limits: Low-volume archived antibody can still be informative, but scarce material reduces your ability to repeat digests, challenge ambiguous calls, or sort out contamination concerns.
Controls and repeat expectations: If an older lot, prior digest, isotype record, or archived MS file exists, keep it available. Comparative evidence can help separate true sequence variation from handling artifacts. Teams should also expect that some sites will need repeat review rather than a one-pass answer.
Batch or contamination risk: Legacy supernatant-derived preparations, shared purification runs, or partially degraded inventories can produce mixed signals. In those cases, weak chain assignment may reflect sample composition rather than interpretation alone.
Interpretation boundaries: A proposed sequence is not proof of functional identity. MS/MS-based reconstruction can leave uncertainty around ambiguous residues, PTM-heavy regions, and sparse peptide overlaps. Database-search support may strengthen conserved regions, but it does not remove uncertainty in unknown CDR sequence. A protein-derived proposal should be treated as a high-value working hypothesis until orthogonal checks and recombinant testing are finished.
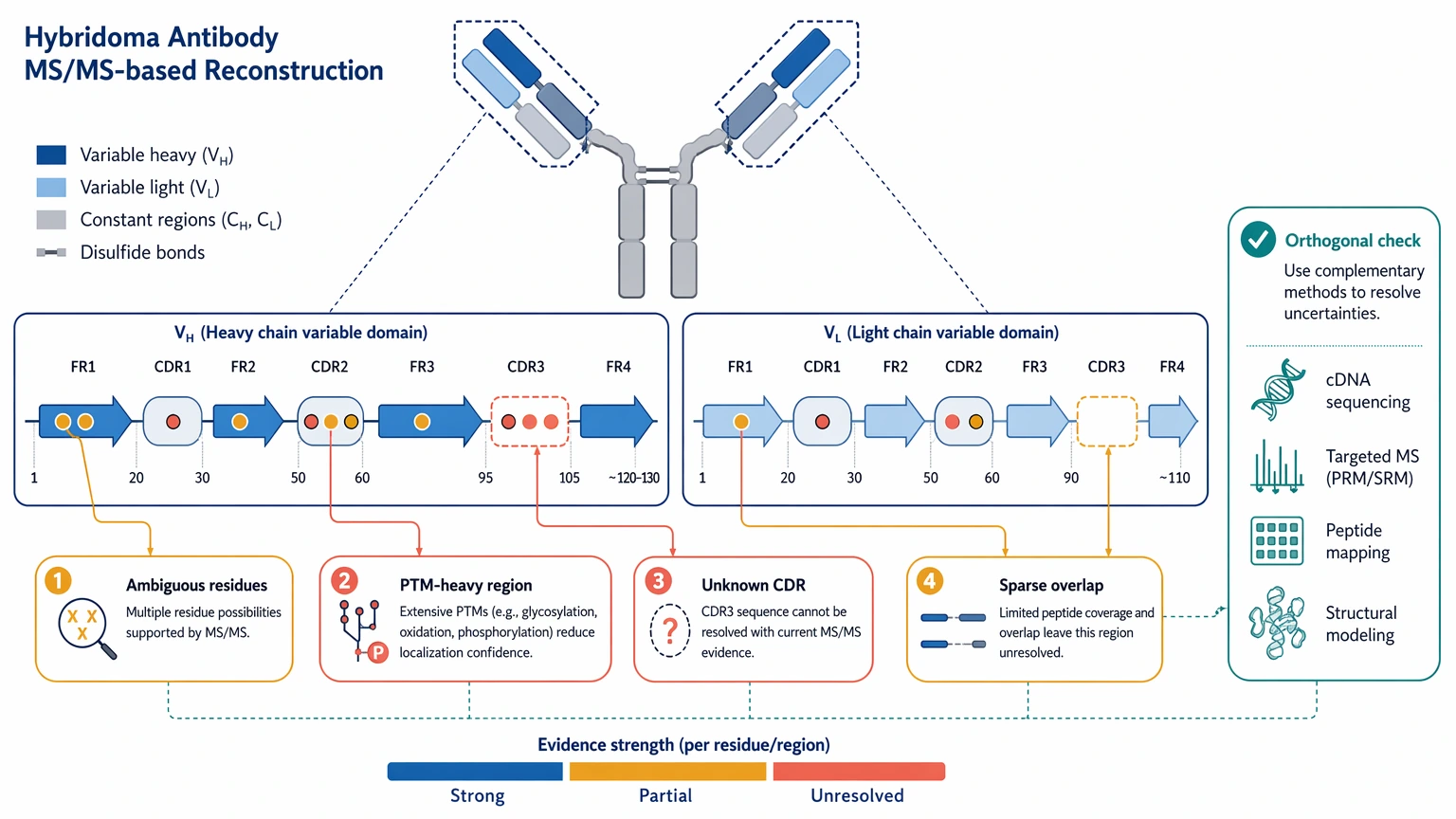
When another method or outside support is the better next step: If the sample is too impure, too degraded, or too limited to support repeat work, a different rescue route may be more efficient. That could mean sourcing cleaner purified antibody, revisiting hybridoma recovery if that is still possible, or using outside analytical support to decide whether a limited protein-first project is technically justified before scarce material is committed.
FAQ
Can purified antibody alone support hybridoma antibody sequencing?
Often, yes. Purified protein can be enough for LC-MS/MS planning if the antibody is intact enough, concentrated enough, and not overwhelmed by contaminants or formulation interference. The practical question is whether the sample can produce overlapping peptide evidence across both chains.
Why is database matching not enough for some hybridoma rescue projects?
Database matching works best when the sequence is already close to a known reference. In rescue projects, the variable region may be undocumented, partially incorrect, or missing from available records. That is why de novo sequencing becomes central.
Which uncertainty matters most before recombinant re-expression?
Uncertainty in CDR-containing regions usually matters more than a small gap in a conserved framework segment. Clone selection gets riskier when functional regions still contain unresolved residue calls.
Can PTMs be mistaken for sequence differences?
Yes. Oxidation, deamidation, pyroglutamate formation, clipping, and glycan-related heterogeneity can shift mass in ways that complicate residue calling. PTM-aware review is needed before a mass shift is treated as sequence evidence.
What information should a team prepare before asking for feasibility review?
Prepare the sample type, available amount, concentration, buffer composition, known isotype, any prior MS or digest files, and a short statement of how the recovered sequence will be used. If you want to evaluate your project, contact MtoZ Biolabs with that material summary and the planned recombinant re-expression context so the workflow can be scoped around the actual decision point.
How to order?

