





How to Use Mass Spectrometry Data for De Novo Sequencing: A Practical Workflow for Unknown Peptide Identification
- a PTM,
- terminal truncation or extension,
- a neutral loss-related fragment pattern,
- unusual cleavage behavior, or
- an unresolved isobaric residue assignment.
- Incomplete ladders can support partial sequence inference without supporting full residue assignment.
- Isobaric residues may remain unresolved.
- PTM localization may stay provisional if site-defining fragments are missing.
- Mixed spectra can make an otherwise plausible sequence difficult to defend.
- Replicate consistency strengthens confidence, but it does not replace fragment-level evidence.
Start de novo sequencing using mass spectrometry data by checking the precursor ion assignment, charge state, monoisotopic mass, and MS/MS spectrum cleanliness before calling any residues. If those upstream assignments are off, every later sequence tag, b-ion and y-ion interpretation, and candidate sequence ranking becomes harder to defend.
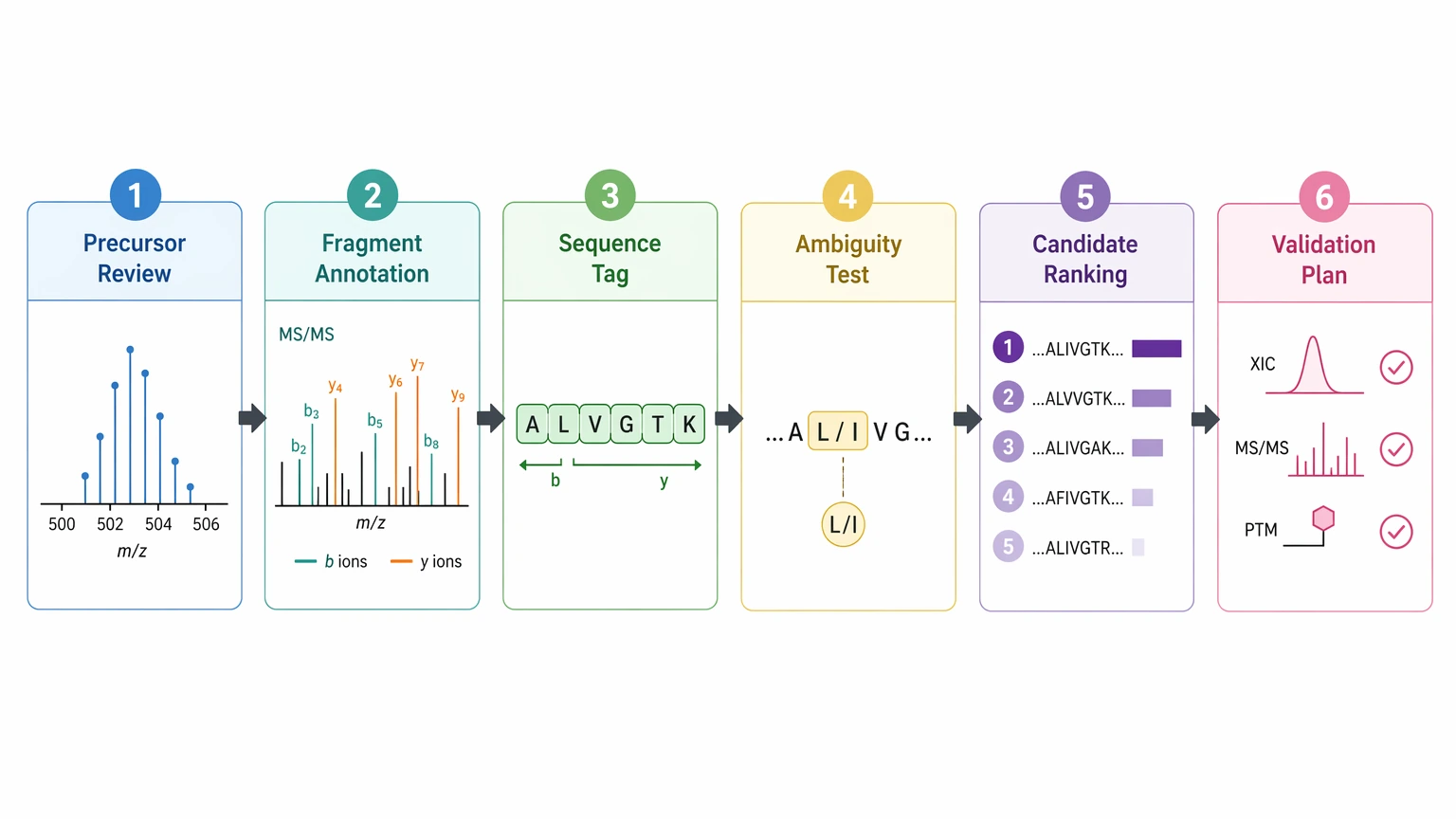
For unknown peptide identification, the most defensible path is precursor review, fragment annotation, sequence tag construction, ambiguity testing, candidate sequence ranking, and orthogonal validation planning. If the fragment evidence does not support a full sequence, do not force one. Report a working candidate sequence with an ambiguity map, show which positions are backed by fragment ion evidence, and state what added LC-MS/MS, alternate fragmentation, purification, or targeted confirmation would make the assignment stronger.
Where the Problem Usually Appears
This issue usually appears after a team detects an unknown peptide feature in LC-MS/MS from a purified fraction, enriched isolate, degradation product, impurity study, venom sample, or modification-rich mixture. A database search may return no confident hit, only a short sequence tag, or several conflicting matches that do not agree with the intact or inferred peptide mass. That is usually the point where database search limitation becomes clear and de novo sequencing becomes the more useful route.
The warning signs in the MS/MS spectrum are usually practical rather than subtle. You may see plausible b ions or y ions, but the ladder breaks. Several major peaks stay unexplained. A mass gap points to a post-translational modification (PTM), truncation, or chemical change, yet the fragment pattern does not localize it cleanly. The real question is not whether software can suggest a sequence. It is whether the data support an assignment that another analyst could review and defend.
That distinction matters because a weak residue call can redirect synthetic peptide confirmation, impurity follow-up, or biological testing toward the wrong target. A disciplined workflow separates well-supported residue calls from unresolved positions as early as possible.
The Most Relevant Root Causes of Interpretation Failure
In this unknown peptide setting, four cause categories explain most interpretation breakdowns.
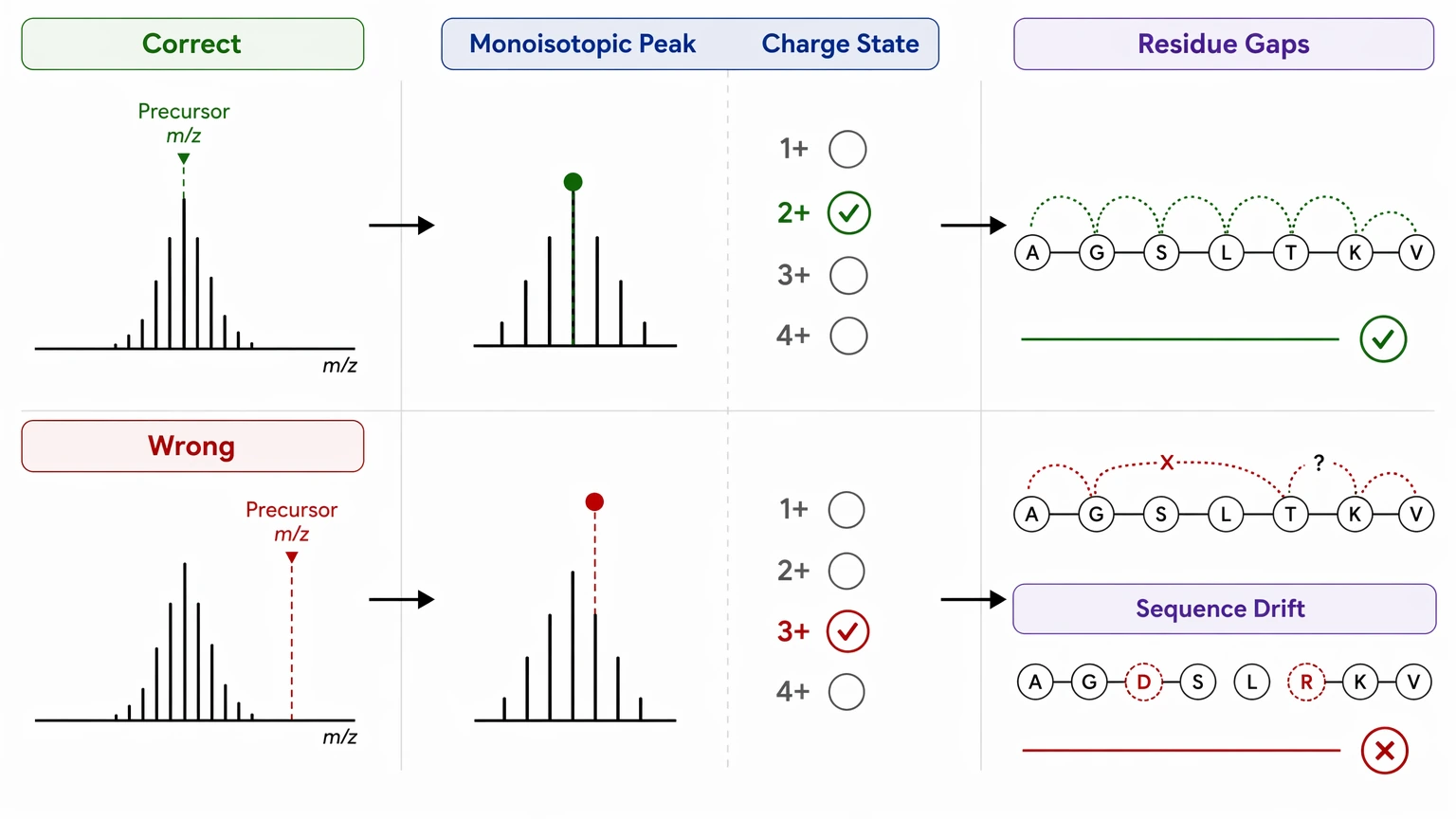
1. Precursor definition errors distort the whole sequence model
If the wrong monoisotopic peak is selected or the charge state is assigned incorrectly, the residue mass gaps downstream will not reconcile. The spectrum may still look partly interpretable, but the sequence logic drifts because the precursor framework is wrong.
2. Fragmentation coverage is too sparse or discontinuous
A useful spectrum does not need a complete ladder, but it does need enough fragmentation coverage to build one or more reliable sequence tags. When long stretches of the b ions or y ions are missing, the data may support only a partial sequence.
3. PTMs or other mass shifts interrupt simple residue calling
Unexpected mass offsets often explain why a ladder stops making sense. Oxidation, amidation, truncation, cyclization, and other changes can alter expected fragment masses. These shifts should be treated as interpretation features rather than dismissed as noise.
4. Mixed or chimeric spectra create false sequence continuity
Poor precursor isolation purity can merge fragments from more than one species into one MS/MS spectrum. That can create misleading ladders, conflicting complementary ions, and candidate sequences that explain only part of the evidence.
A Practical Data-Interpretation Workflow for Unknown Peptide Identification
This is a data-interpretation problem, so the workflow should focus on reading evidence in the right order rather than restating an entire proteomics pipeline.
Step 1: Confirm precursor-level evidence before annotating fragment ions
Start with the precursor ion. Record the precursor m/z, charge state, isotope pattern, retention behavior, and monoisotopic mass assignment. Then confirm whether the isotope envelope supports the assigned charge state, whether the precursor fits the expected peptide mass range, and whether isolation was clean enough to reduce co-isolation. If any of those points remain uncertain, re-extract or reprocess the data before doing detailed spectral annotation.
Step 2: Decide whether the MS/MS spectrum is interpretable enough
A quick screening table helps separate a taggable spectrum from one that still needs better acquisition or cleaner precursor selection.
| Checkpoint | What to Look For | Why It Matters |
|---|---|---|
| Fragmentation coverage | b ions and/or y ions across multiple positions | Supports residue-by-residue reading |
| Signal pattern | Annotatable peaks above background | Reduces false assignments |
| Complementary evidence | Some matched b/y relationships | Improves sequence confidence |
| Mass error | Consistent fragment matching tolerance | Supports credible annotation |
| Unexplained major peaks | Limited or localized | Keeps ambiguity manageable |
If most intense peaks remain unexplained, or if the ladder appears only as isolated fragments, the spectrum may support a sequence tag but not a complete candidate sequence.
Step 3: Build sequence tags from the clearest ion ladder
Start from the most continuous series, not from the terminus you expect to see. In many unknown peptide datasets, a central stretch of fragments is clearer than either end. Translate mass differences between consecutive fragment ions into residue candidates and mark each uninterrupted run as a sequence tag.
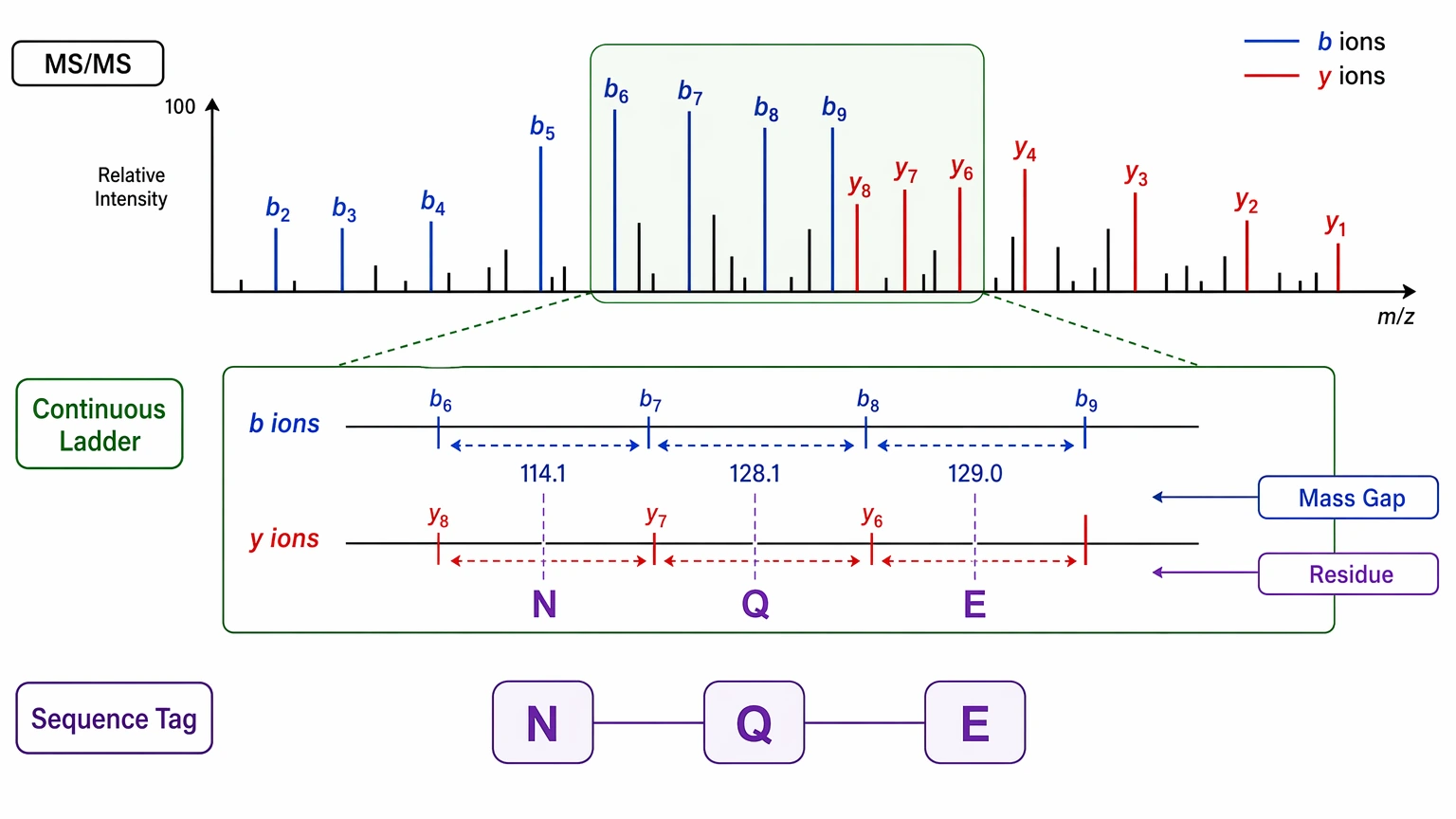
This step works best when you prioritize the ladder with the fewest internal contradictions, check whether mass gaps fit expected amino acid increments, label unresolved positions instead of forcing a call, and compare b-ion and y-ion evidence wherever both are present. This is also where leucine/isoleucine ambiguity needs careful handling because those isobaric residues often remain indistinguishable by mass alone in routine tandem mass spectrometry.
Step 4: Test PTM and ambiguity hypotheses where the ladder breaks
Once you have a first-pass tag, focus on the breakpoints. Ask what the unexplained mass gap might represent:
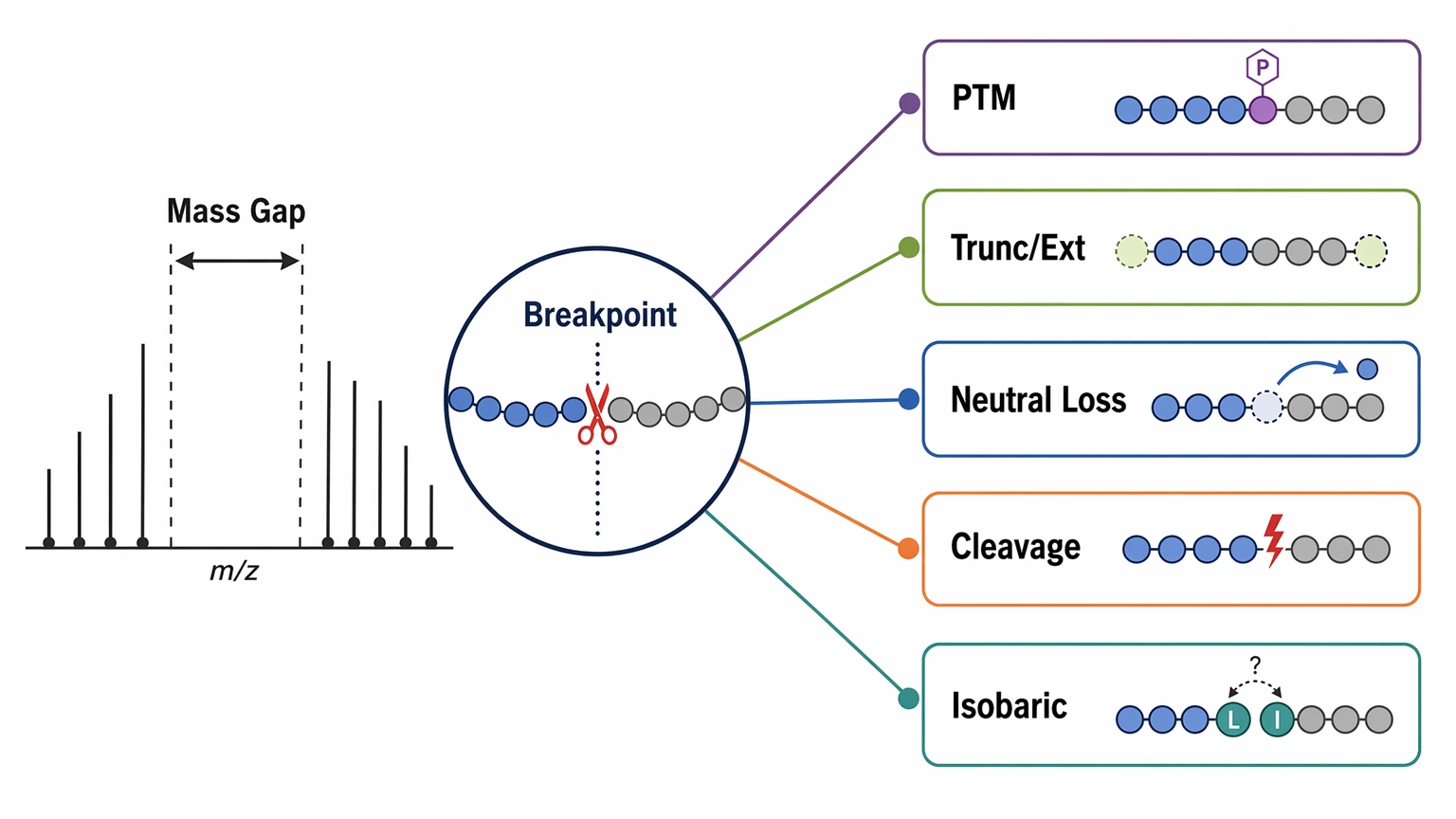
A localized mass offset with supporting neutral loss behavior can support a modification hypothesis, while scattered mismatches across the spectrum more often point to mixed spectra or incorrect precursor selection. If your team is already working with a partial tag, modification-rich spectrum, or mixed-spectrum question, you can submit your requirements to MtoZ Biolabs to evaluate your project against a de novo peptide sequencing or LC-MS/MS interpretation workflow.
Step 5: Rank candidate sequences against the whole spectrum
Once one or more candidate models exist, rank them against the full dataset rather than one local ladder. Useful criteria include how many major fragment ions each model explains, whether the mass error pattern stays coherent, whether complementary ions support the same sequence logic, how many dominant peaks remain unexplained, and whether ambiguities are localized or spread across the peptide. A strong candidate sequence does not require certainty at every position, but it does require transparent evidence.
Step 6: Choose the most informative next action
At this stage, the decision is practical: report now, reacquire data, or move into targeted confirmation.
| Situation | Best Next Action |
|---|---|
| Good ladder continuity with limited ambiguity | Report the candidate sequence with an evidence summary |
| Strong partial sequence tag but missing internal coverage | Reacquire MS/MS or use alternate fragmentation |
| Suspected PTM with weak localization | Add targeted follow-up or modification-focused review |
| Signs of chimeric spectra | Purify further or tighten precursor isolation |
| Two or more near-equal candidates | Plan orthogonal validation before final reporting |
The output from this step should state whether the current dataset supports a reportable candidate sequence, a partial tag with open ambiguity, or a follow-up experiment request.
Service Routes to Consider
When a project moves from partial sequence recovery to confirmation planning, the most useful service routes are the ones that match the current bottleneck in interpretation rather than repeating the same acquisition step.
What a Defensible Output Should Look Like
A useful de novo result is rarely just a single-line sequence claim. The more realistic deliverable is a compact evidence package that includes the confirmed precursor definition, a summary of spectral annotation, one or more sequence tags, a ranked candidate sequence list, an ambiguity map, PTM hypotheses where relevant, and recommended orthogonal validation steps. That format shows which residues are well supported and which still need confirmation.
Follow-Up Validation That Actually Matches the Weak Point
Validation should target the specific uncertainty in the dataset. If the main issue is weak PTM localization, targeted MS follow-up may be enough. If the main issue is mixture complexity, another round of interpretation on the same mixed sample usually will not solve it. If the unresolved site reflects leucine/isoleucine ambiguity, repeated routine MS/MS may still leave the same ambiguity in place. Useful orthogonal validation options include targeted MS confirmation, synthetic peptide comparison, additional purification, alternate fragmentation modes such as ETD or EThcD when appropriate, or N-terminal work when the peptide chemistry allows it.
Common Boundaries to State Clearly in the Report
Conclusion
De novo sequencing using mass spectrometry data is most credible when the assignment is built from precursor accuracy, fragment continuity, sequence tag logic, and explicit ambiguity handling rather than a forced full-sequence guess. For unknown peptide identification, the strongest workflow is to confirm the precursor ion first, annotate the clearest b ions and y ions, rank each candidate sequence against the full MS/MS spectrum, and use orthogonal validation when PTMs, chimeric spectra, or residue ambiguity still limit sequence confidence.
This workflow fits projects involving purified unknown peptides, impurity characterization, degradation products, and modification-rich analytes where database search limitation blocks a confident identification. If your team has a partial sequence tag, unresolved mass shifts, or several competing sequence models, contact us at MtoZ Biolabs to discuss the dataset, submit your requirements, and plan the most suitable confirmation path before spending more sample on follow-up work.
FAQ
Can I do de novo sequencing if database search returns a weak partial hit?
Yes. A weak database hit can still provide context, especially if it suggests homology, truncation, or a likely residue motif, but it should not override fragment-level contradictions.
Which fragmentation mode is usually most helpful for unknown peptide identification?
HCD or CID often gives useful b/y ladders, while ETD or EThcD may help when labile modifications or charge-rich peptides limit conventional fragmentation.
What peptide length is hardest to interpret by de novo sequencing?
Very short peptides may not generate enough distinguishing fragment evidence, while longer peptides often show broken ladders and multiple unresolved regions.
How many unexplained peaks are too many?
No fixed cutoff is reliable. The practical question is whether the unexplained peaks are minor background features or dominant signals that compete with the proposed annotation.
Is manual interpretation still useful when software already proposes sequences?
Yes. Manual review is often needed to confirm precursor assignment, inspect sequence tags, recognize mixed spectra, and judge whether a PTM hypothesis fits the actual fragment evidence.
What should I include when sending an unknown peptide dataset for external review?
Provide raw or processed LC-MS/MS files, precursor m/z, charge-state information, sample source, purification status, suspected PTM context, and any current sequence tags or candidate sequences.
Related Services
Main Service |
Supporting Service |
Validation Service |
Alternative Service |
How to order?

