





How to Scope a Peptide De Novo Sequencing Project: Sample Type, Spectral Readiness, and Deliverable Planning
- an unknown HPLC peak that does not match any expected component
- a bioactive peptide fraction from a natural source or fermentation workflow
- an impurity or degradation product in a peptide development program
- a novel engineered peptide outside the reference database
- an LC-MS/MS dataset with weak database-search hits but no clear follow-up plan
- support synthetic peptide design for a novel analyte
- determine whether an impurity is a truncation or a modified form
- triage archived LC-MS/MS data before ordering new acquisition
- decide whether a PTM-rich peptide can support a consensus sequence or only a sequence tag
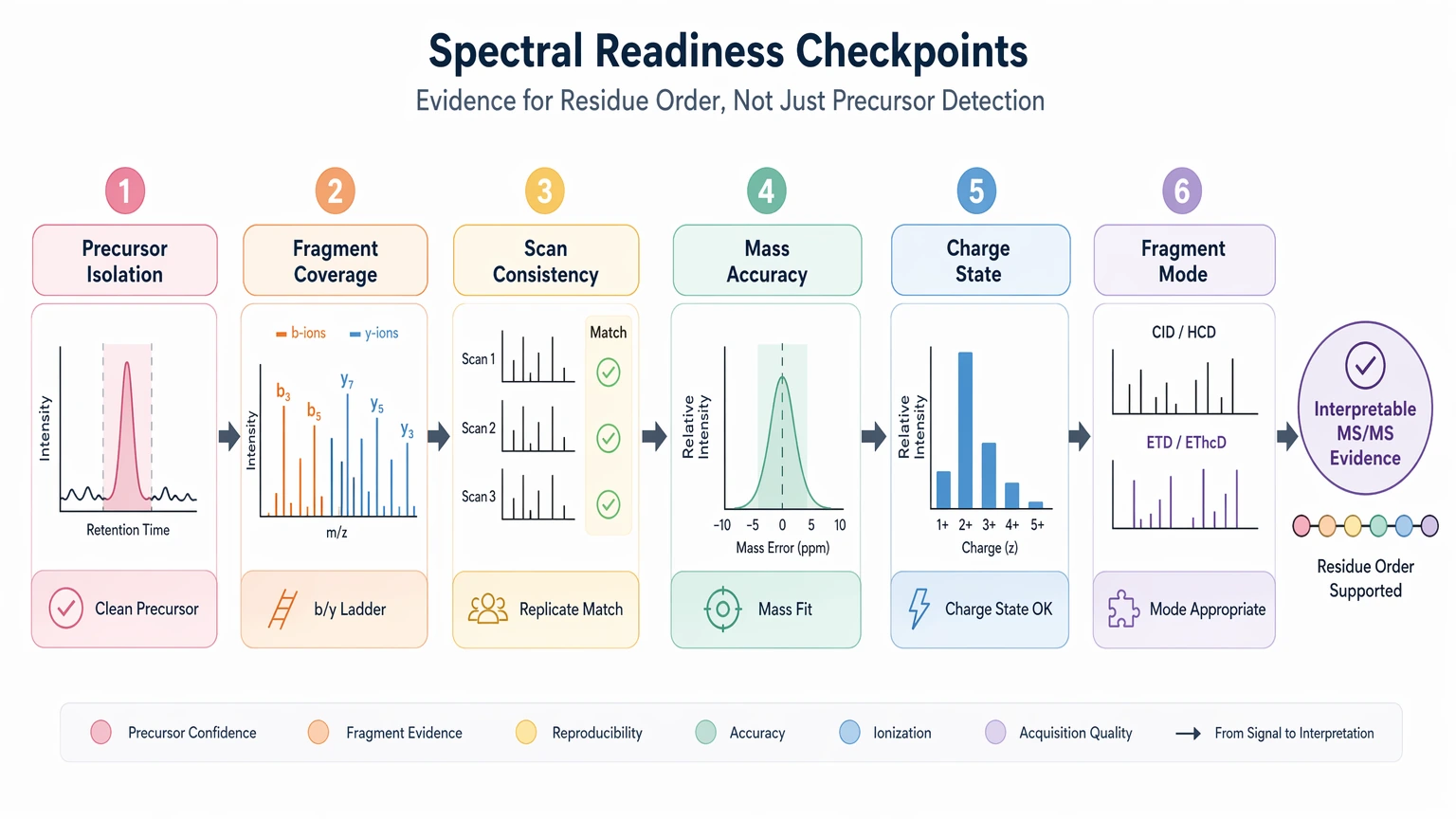
- precursor isolation: is the target precursor ion cleanly isolated or likely co-isolated with nearby species?
- fragmentation coverage: do b ions, y ions, or complementary fragment ion series support a readable ladder?
- spectral quality across scans: are informative fragment ion patterns reproducible across scans or replicates?
- mass accuracy: does the reconstructed path remain consistent with precursor mass and intact mass information?
- charge state behavior: does the observed charge state support informative fragmentation?
- fragmentation mode availability: if PTM localization or labile features matter, is complementary fragmentation available?
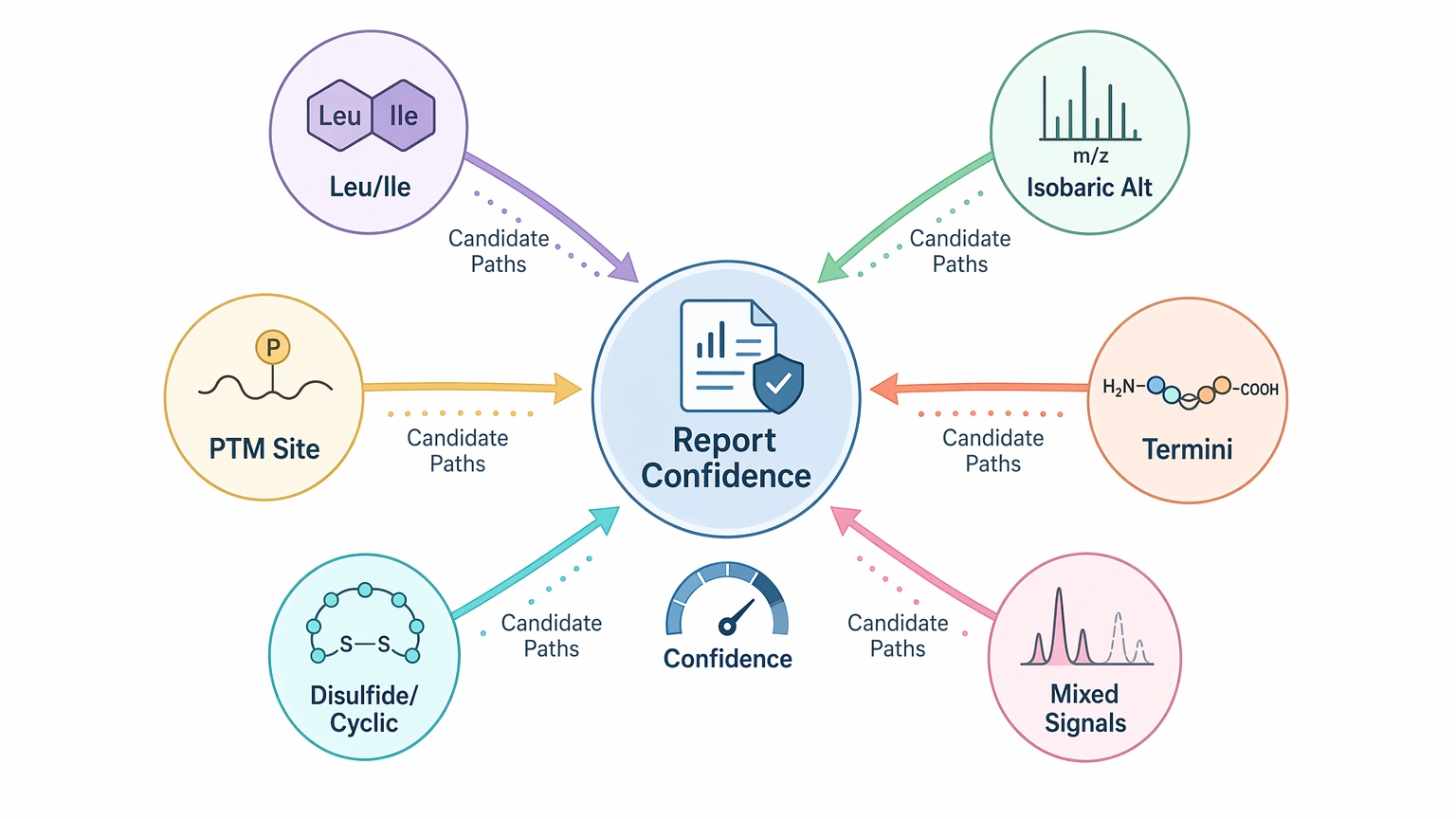
- leucine/isoleucine ambiguity
- isobaric residue or modification alternatives
- post-translational modification (PTM) localization uncertainty
- terminal modification or blocked termini
- disulfide bond or cyclic peptide complexity
- mixed peptide population and overlapping fragment evidence
-
Raw data review only
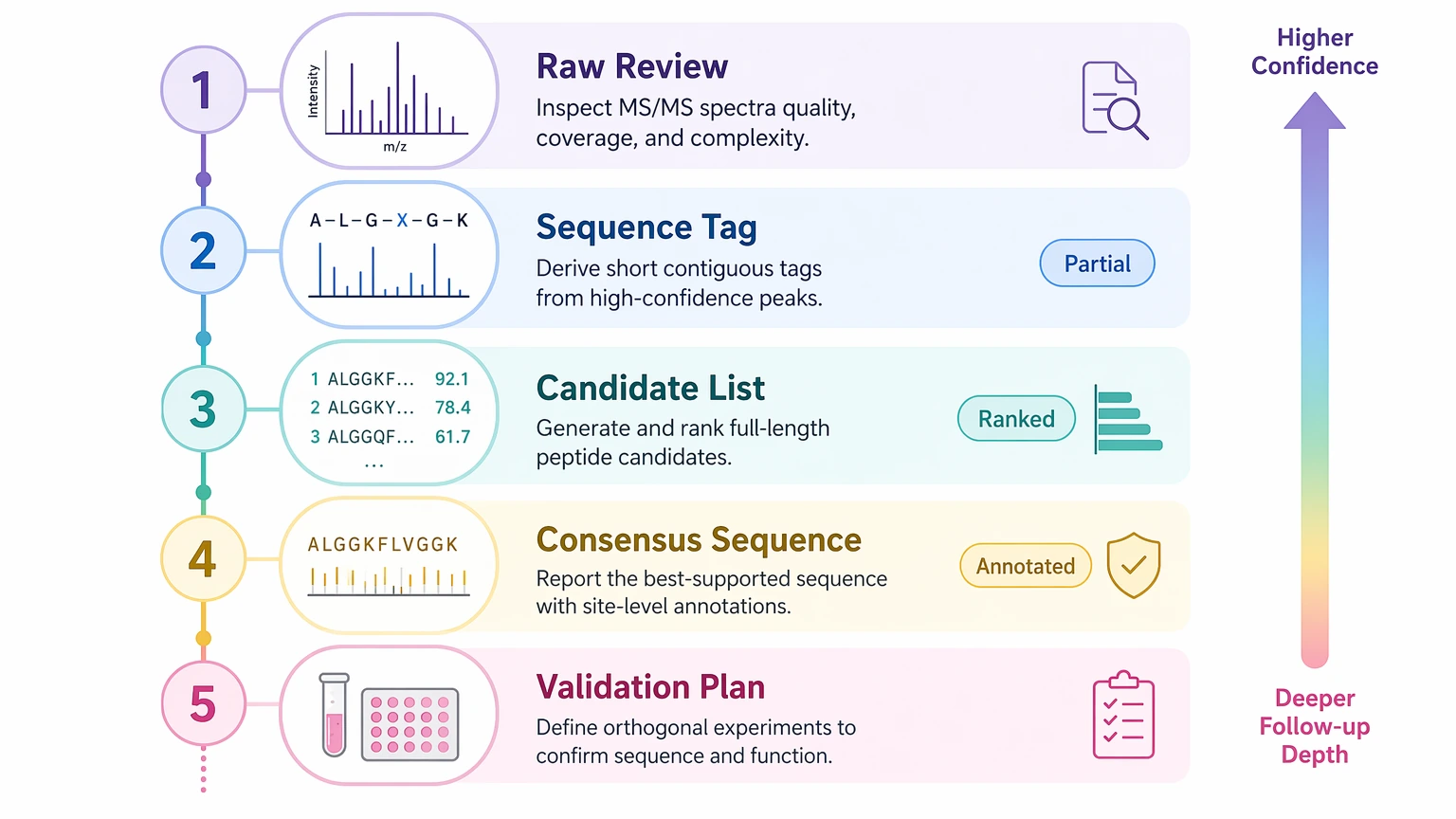
For archived files that first need a readiness decision. -
Sequence tag
For partial but still useful residue-order information. -
Ranked candidate sequence list
For cases where more than one candidate sequence matches the evidence. -
Consensus sequence with confidence annotation
For broader residue ordering with clearly marked uncertain positions. -
Sequence-plus-validation plan
For synthesis support, impurity work, or function-driven follow-up. - candidate sequence or consensus sequence output
- annotated spectrum figures
- residue-level confidence annotation
- PTM annotation status and PTM localization limits
- unresolved-position commentary
- intact mass reconciliation
- orthogonal validation recommendations
- whether the current sample and spectra are suitable for de novo peptide sequencing
- whether the evidence supports a sequence tag, candidate sequence list, or consensus sequence
- which positions or modifications remain conditional
- how annotated spectrum evidence supports each residue call
- intact mass cross-checking
- targeted LC-MS/MS confirmation of critical transitions
- synthetic peptide comparison against a candidate sequence
- repeat acquisition with improved precursor isolation
- escalation to de novo protein sequencing when the analyte behaves more like a small protein than a peptide
- Sample quality or amount limits: low material, high salt, detergents, or incompatible buffers can restrict cleanup, repeat injections, and complementary fragmentation.
- Controls and repeat expectations: if contamination is plausible, document purification history, neighboring fractions, blank performance, and whether repeat analysis is possible.
- Batch or contamination risk: co-eluting species and carryover can distort precursor isolation and create misleading fragment ion evidence.
- Interpretation boundaries: a top-ranked candidate sequence is not automatically a fully confirmed structure. Sequence confidence may remain local rather than global, especially when PTMs, terminal modification, or isobaric residue assignments are involved.
- When another method or outside support is better: if the sample is a mixed peptide population, the spectrum contains major ladder gaps, or cyclic peptide and disulfide bond features dominate the problem, further purification, alternate fragmentation, de novo protein sequencing, or outside expert review may be the better next step.
Move a de novo peptide sequencing project forward only after three points are clear: the sample is tractable enough to isolate a real target, the LC-MS/MS data contain interpretable fragment ion evidence, and the requested output fits the downstream decision. If even one of those points is still vague, the better next move is usually to narrow the scope before starting full sequence interpretation.
Quick decision block
Proceed now when you have a purified or well-targeted analyte, usable precursor isolation, and a clear need for a candidate sequence, consensus sequence, or validation plan.
Pause and improve the package when the sample is mixed, spectra show weak fragmentation coverage, or PTM localization and terminal modification status are still undefined.
Redefine the objective when the data are more likely to support a sequence tag or ranked candidate sequence list than a single high-confidence sequence.
For most teams, the simplest planning method is to separate sample suitability from spectral suitability. A peptide submission can look clean on paper and still fail de novo peptide sequencing if the precursor ion is not isolated well or the fragment ion ladder is too incomplete to support residue order. On the other hand, archived raw files sometimes already contain enough information for unknown peptide identification before any new material is shipped.
Where Scoping Problems Usually Start
This problem usually shows up before budget signoff or provider discussions. A team may already know that a routine database search did not resolve the analyte, but that alone does not mean de novo peptide sequencing is ready to begin.
Typical triggers include:
The gap is often operational, not theoretical. Teams ask for a “final sequence” before deciding whether they really need synthesis support, impurity attribution, or only early triage. That disconnect can lead to rework, repeat injections, or a report that is scientifically sound but less useful than expected for the actual project decision.
Brief Root-Cause Analysis: Why Projects Get Mis-Scoped
Most readiness issues here fall into four groups.
1. The sample description is too broad
“Peptide sample” can refer to a purified peptide, a mixed peptide population, a digest-derived unknown peptide, or an intact small protein. Those inputs are not interchangeable. Each one changes precursor isolation difficulty, fragmentation behavior, and how realistic it is to obtain a consensus sequence.
2. The spectra are present but not interpretation-ready
A raw file can look acceptable at first glance and still be weak for de novo peptide sequencing. Common limits include low signal-to-noise, incomplete b ions or y ions, uncertain charge state assignment, and poor precursor isolation.
3. Ambiguity is underestimated
Leucine/isoleucine ambiguity, isobaric residue assignments, post-translational modification (PTM) burden, terminal modification, disulfide bond structure, and cyclic peptide architecture can all lower sequence confidence. Even strong tandem mass spectrometry data may support several candidate sequence paths rather than one fully resolved answer.
4. Deliverables are defined too loosely
“Identify the peptide” is not enough as a project scope. The report may need a sequence tag, a ranked candidate sequence list, a consensus sequence with confidence annotation, or a sequence-plus-validation plan. Those are different outputs and should be requested directly.
A Project-Planning Workflow for De Novo Peptide Sequencing
Step 1: Define the decision endpoint before the analytical scope
Write one sentence that states what the project must decide. Good examples include:
This step keeps the workflow tied to the real research or business decision. A ranked candidate sequence may be enough for discovery triage, while impurity attribution or synthesis usually needs stronger confidence annotation and follow-up confirmation.
Step 2: Triage the sample type separately from the data package
Use the sample form and the available evidence as two separate planning inputs.
| Sample type | Best fit | Main constraint | Practical next step |
|---|---|---|---|
| Purified peptide peak | Full de novo peptide sequencing | Hidden co-elution can still remain | Submit sample and prior raw files together |
| Mixed fraction | Feasibility review or sequence tag recovery | Mixed peptide population complicates precursor isolation | Purify further or narrow the scope |
| Intact small protein | De novo protein sequencing or hybrid strategy | Larger sequence space and disulfide bond complexity | Confirm whether peptide-level or protein-level output is needed |
| Archived LC-MS/MS raw data only | Spectral readiness review | No chance to improve acquisition if the file is weak | Request raw file triage first |
| Digest-derived unknown peptide | Targeted unknown peptide identification | Background peptides may interfere | Confirm precursor uniqueness |
A purified sample with enough material for repeat analysis is the most favorable starting point. A mixed fraction with no replicate capacity gives you much less room for error.
Step 3: Check spectral readiness with de novo-specific criteria
De novo interpretation depends on evidence for residue order, not just precursor detection. Review the LC-MS/MS data with the following checkpoints:
The table below organizes Evidence, What it supports, Main limitation so the project decision can be checked before the next workflow step.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
| Evidence | What it supports | Main limitation | Follow-up |
|---|---|---|---|
| Clean precursor isolation | Target-focused interpretation | Does not ensure full fragment coverage | Check ladder continuity |
| Continuous b ions or y ions | Residue order across part of the peptide | Gaps can leave multiple candidate sequence paths | Mark unresolved positions |
| Similar replicate spectra | Better confidence annotation | Does not remove leucine/isoleucine ambiguity | Plan orthogonal validation |
| Precursor/reconstruction mass agreement | Internal consistency | PTMs or terminal modification may still fit more than one model | Compare alternatives |
One limitation is worth stating early: even when the LC-MS/MS data are strong, standard MS/MS interpretation does not always resolve leucine/isoleucine ambiguity, PTM localization, or every database-search limitation with a single unqualified sequence call.
Step 4: List the main ambiguity drivers before you request a final output
Before kickoff, identify which uncertainty sources are most likely to shape the report:
This is also the point where a consultation can save time. If the project may require re-acquisition, orthogonal fragmentation, or a narrower interpretation target, submit your requirements and evaluate your project before locking in the final report format.
Step 5: Request deliverables in analytical terms
A practical de novo peptide sequencing scope usually fits one of five output levels:
At minimum, ask for:
If your team is deciding between a quick feasibility review and a full interpretation package, contact MtoZ Biolabs to submit your requirements and evaluate your project scope before the report format is fixed.
Expected Results and Validation Methods
A well-scoped project should first produce decision-grade clarity, then a validation path.
Immediate deliverables
The first report should usually establish:
Follow-up confirmation
The next layer is confirmation, not repetition of the same interpretation. Depending on the use case, follow-up confirmation may include:
If your downstream decision depends on synthesis, impurity assignment, or a stronger sequence claim, contact MtoZ Biolabs to submit your requirements and evaluate your project around both de novo interpretation and orthogonal validation rather than treating the sequence report as the only endpoint.
Key Cautions and Practical Limits
A realistic scope should also define what could block or weaken interpretation.
Conclusion
A de novo peptide sequencing project is ready when sample purity, spectral quality, and deliverable expectations all line up. In practice, that means deciding whether you have a tractable unknown peptide, whether the LC-MS/MS evidence supports sequence reconstruction rather than only precursor detection, and whether the report should end with a sequence tag, candidate sequence set, or consensus sequence plus confirmation plan.
This framework is especially useful for unknown peptide identification, impurity characterization, natural-source peptide discovery, and archived data triage when a routine database search leaves open questions. If your team needs to align sample submission, raw-data review, and validation planning before approval, contact MtoZ Biolabs to discuss the sample or dataset and evaluate the project scope against the decision you actually need to make.
FAQ
Can I submit raw files first and ship material later?
Yes. A raw-file triage step is often the fastest way to judge spectral readiness before using up limited sample. It can show whether the current data support full interpretation or whether re-acquisition should come first.
How pure does the sample need to be before de novo work is realistic?
There is no single cutoff for every analyte. The practical test is whether the target precursor ion can be isolated cleanly enough to generate interpretable fragment ion evidence. A dominant peak can still contain co-eluting species.
When is a ranked candidate sequence list acceptable?
It usually works for early triage, shortlist generation, or hypothesis narrowing. It is a weaker fit when the result will be used for synthesis, formal impurity assignment, or structure-sensitive biological follow-up.
Should I request PTM localization in the first report?
Request it when modification status affects the decision, but ask for localization confidence to be stated explicitly. A mass shift may be supported even when the exact PTM site remains conditional.
What project details save the most time during consultation?
The most useful inputs are precursor m/z, charge state, estimated sequence length, sample origin, purification history, suspected PTMs, intact mass if available, and the exact downstream use of the result.
When does this become a de novo protein sequencing project instead of a peptide project?
That shift usually happens when the analyte behaves like an intact small protein, includes more complex disulfide bond patterns, or needs broader sequence reconstruction than peptide-level interpretation can reasonably support.
How to order?

