





Efficient and Accurate Protein Sequencing Methods: Mass Spectrometry, Edman Degradation, and Workflow Selection
-
Mass spectrometry-based protein sequencing is the main route for efficient analysis of complex or low-abundance samples.
-
Edman degradation is slower and more limited in throughput, but it remains valuable for clean N-terminal confirmation.
-
De novo sequencing becomes important when reference databases are incomplete or when novel variants are expected.
-
Terminal sequencing, intact mass analysis, and PTM-aware workflows often complement rather than replace standard LC-MS/MS sequencing.
-
For discovery-scale projects, LC-MS/MS usually gives the best balance of throughput and sequence information.
-
For clean terminal questions, Edman or dedicated terminal analysis can be faster to interpret than a full proteomics workflow.
-
For unknown sequences, de novo support prevents overreliance on incomplete databases.
-
For modified or heterogeneous proteins, combining intact mass, peptide mapping, and targeted confirmation often improves accuracy.
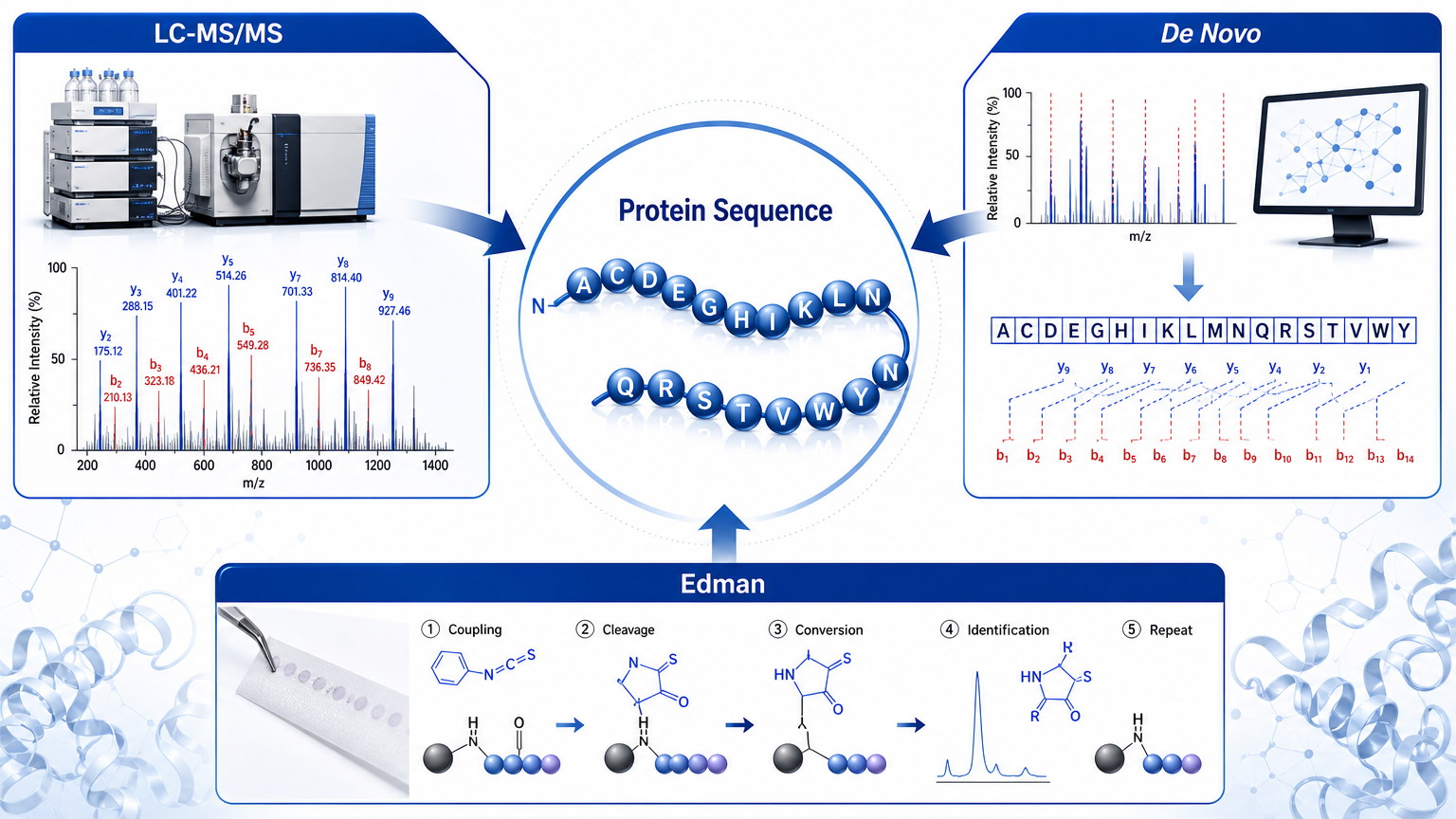
Efficient and accurate protein sequencing depends on matching the method to the sample, the biological question, and the level of sequence detail required. In modern workflows, mass spectrometry is usually the most scalable option because it can identify peptides and proteins in complex mixtures, while Edman degradation and terminal sequencing remain useful when clean sequence confirmation is needed at a defined protein end.
Key Takeaways
What Does Protein Sequencing Really Mean?
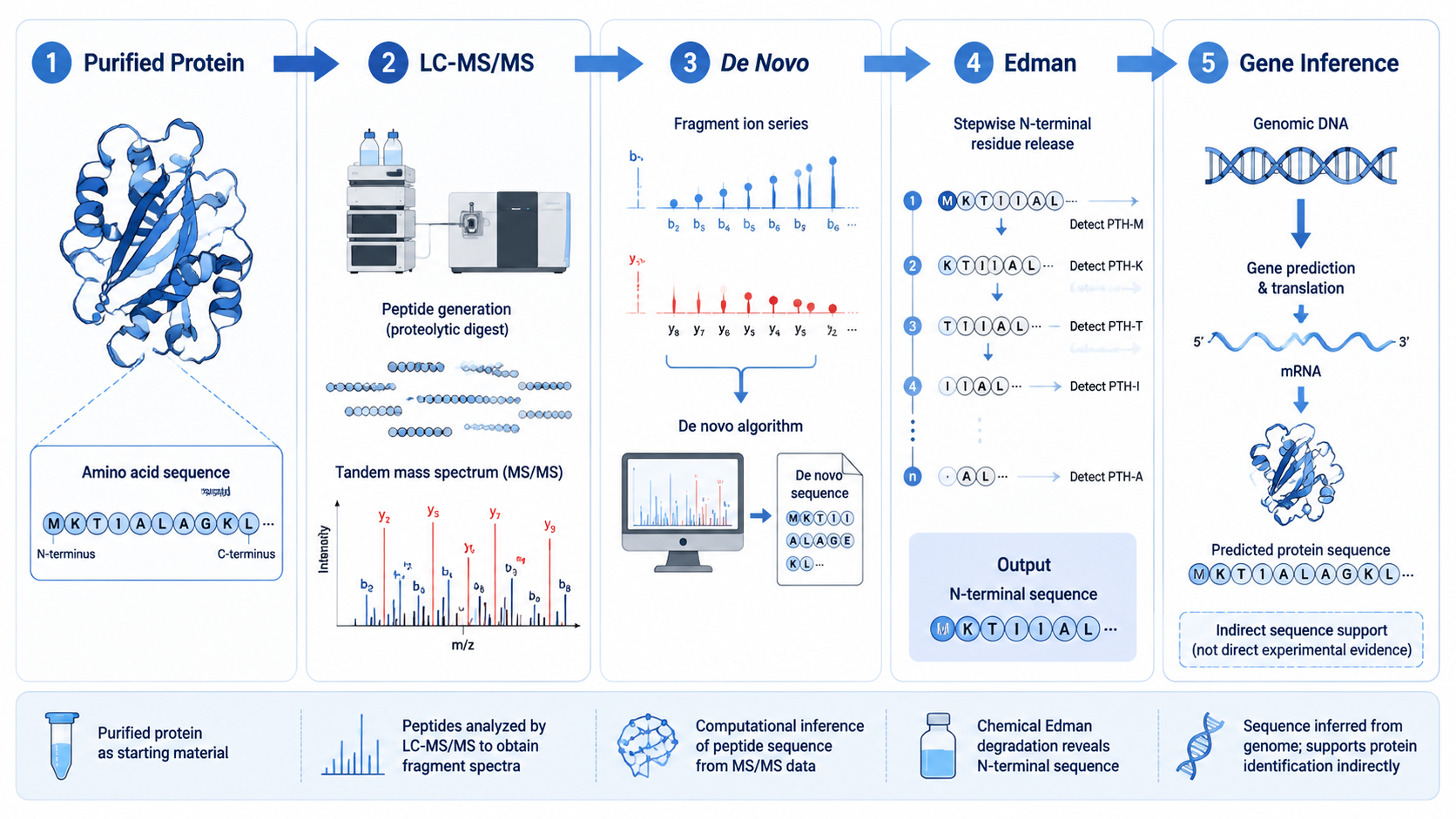
Protein sequencing can refer to several different analytical tasks: confirming the amino acid order of a purified protein, reconstructing peptide sequences from tandem mass spectra, identifying unknown proteins from peptide evidence, or inferring expected protein sequence from genomic or transcriptomic information.
Techniques such as Sanger sequencing or next-generation sequencing do not sequence proteins directly. They sequence nucleic acids and can help predict a protein sequence only when gene or transcript information is the right proxy.
Related Services
Protein Sequence Analysis Service
Mass Spectrometry Protein Sequencing Service
Edman Based Protein Sequencing Service
Main Methods for Efficient and Accurate Protein Sequencing
1. LC-MS/MS and Peptide-Based Sequencing
LC-MS/MS is the default choice for modern protein sequencing projects because it combines sensitivity, throughput, and flexibility. Proteins are digested into peptides, peptides are separated by liquid chromatography, and tandem mass spectra are collected so fragment ions can be matched to database sequences or interpreted de novo.
2. De Novo Peptide Sequencing
De novo sequencing uses fragment ion spacing to infer peptide sequence without relying entirely on a reference database. It becomes essential when unknown variants, antibodies, toxins, microbial peptides, engineered proteins, or sequence-divergent targets are involved.
3. Edman Degradation
Edman degradation removes one residue at a time from the N terminus of a purified peptide or protein and identifies the released amino acid. The method is accurate for clean samples, but it is not efficient for complex mixtures or long proteins that require broad sequence coverage.
4. N- and C-Terminal Sequencing
Terminal sequencing methods are often used to confirm processing sites, leader peptide removal, truncation, mature protein boundaries, or biopharmaceutical product integrity.
5. Gene- or Transcript-Based Sequence Inference
When the goal is to predict a protein sequence encoded by a known gene, nucleic acid sequencing can contribute supporting evidence. Still, this is indirect and cannot replace protein-level analysis when researchers need to confirm translation products, cleavage, PTMs, or unexpected heterogeneity.
Why Does Method Choice Matter?
An efficient workflow is not simply the fastest workflow. It is the one that answers the sequence question with the least unnecessary complexity and the highest acceptable confidence.
Strengths and Tradeoffs of Common Methods
| Method | Best for | Main Strength | Main Limitation |
|---|---|---|---|
| LC-MS/MS database search | Known organisms or reference-rich systems | Fast, scalable identification in complex mixtures | Depends on database completeness |
| LC-MS/MS plus de novo | Unknown or sequence-divergent targets | Recovers novel peptide sequence information | Requires stronger spectra and more review |
| Edman degradation | Purified proteins or peptides with accessible N termini | High-confidence stepwise terminal readout | Low throughput and limited depth |
| N-/C-terminal sequencing | Processing-site and boundary confirmation | Direct answer to terminal questions | Narrower scope than full sequence mapping |
| Gene-based inference | Expected coding sequences from known genes | Strong upstream sequence expectation | Indirect and blind to protein-level processing |
Choosing the Right Protein Sequencing Workflow
The first decision is whether the sample is purified or complex. The second is whether a trustworthy reference sequence exists. The third is what level of accuracy the project requires, because some studies need residue-level evidence or terminal confirmation rather than simple identification.
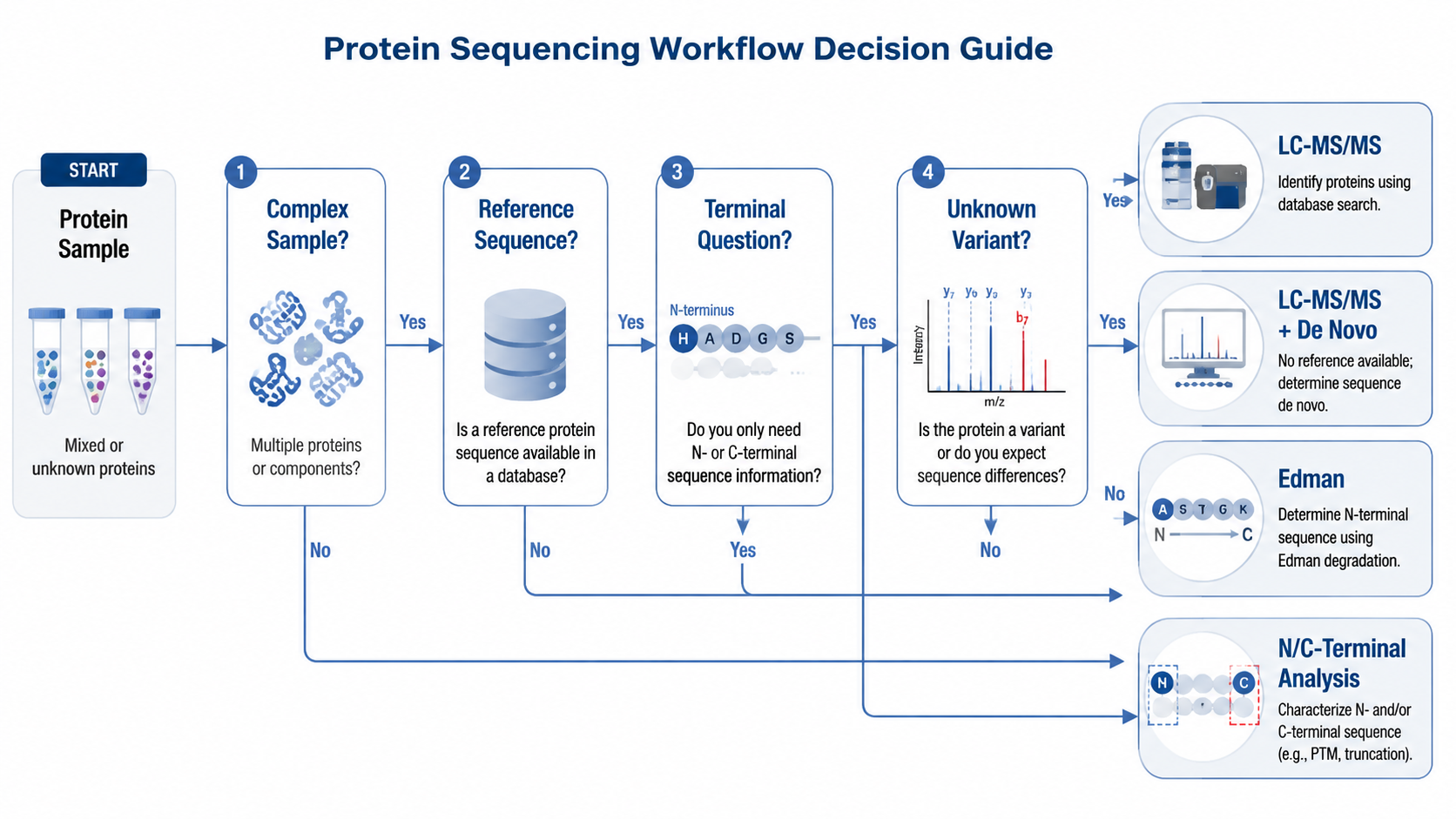
Typical Application Scenarios
1. Discovery Proteomics
Use LC-MS/MS with database search when the main goal is large-scale protein identification in research samples.
2. Novel Peptide or Antibody Analysis
Add de novo sequencing when reference sequences are incomplete or when engineered, immune, or non-model targets may contain unexpected sequence features.
3. Biopharmaceutical QC and Characterization
Combine peptide mapping, terminal analysis, and intact mass information when product identity, clipping, heterogeneity, or processing must be confirmed with high confidence.
4. Targeted Terminal Confirmation
Use Edman or terminal-focused approaches when the key question is the exact N- or C-terminal boundary rather than broad proteome coverage.
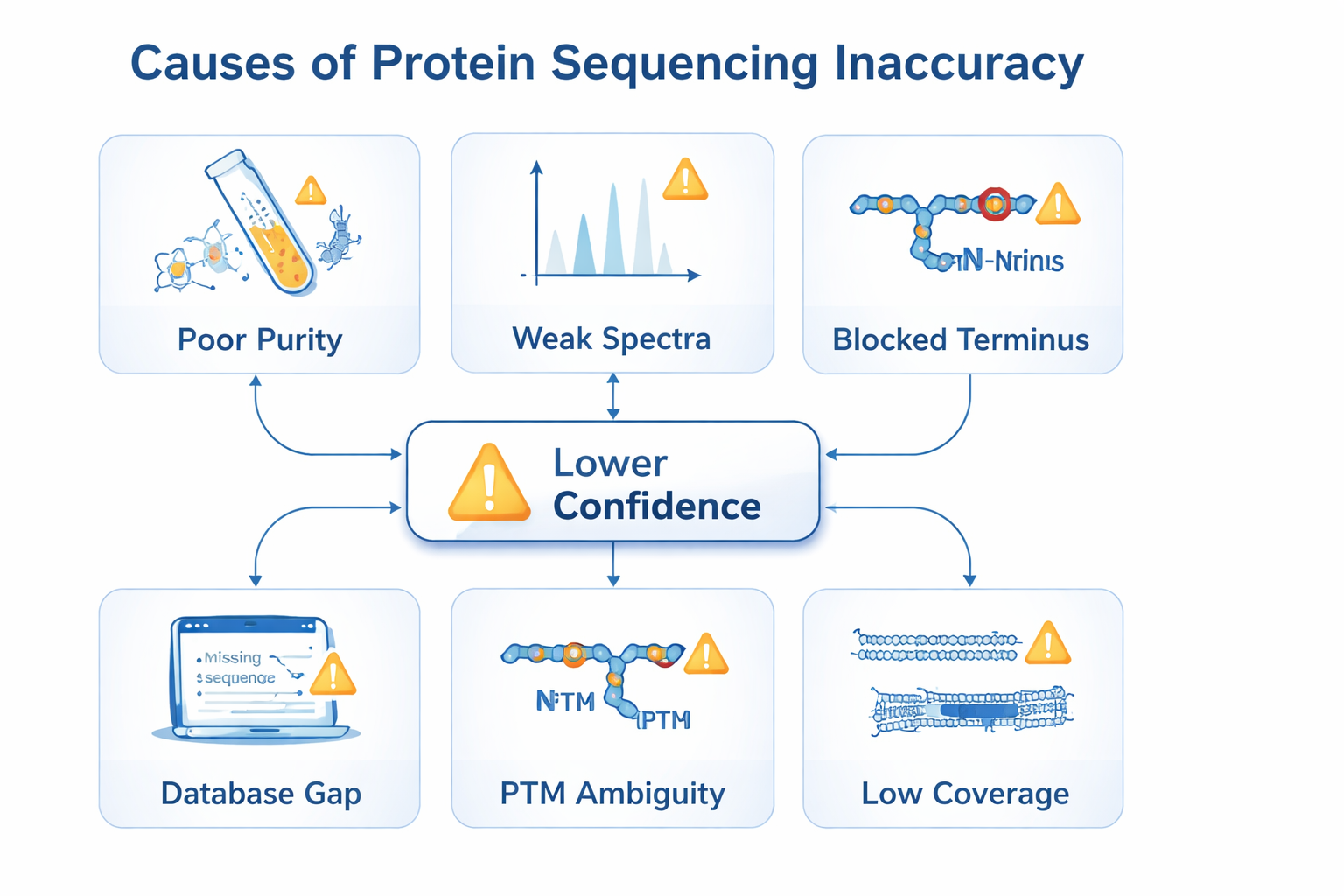
Common Sources of Sequencing Error
Accuracy drops when sample cleanup is poor, sequence coverage is sparse, spectra are weak, modifications complicate fragmentation, or the analysis assumes the wrong reference database. In practice, the safest workflows treat sample preparation, instrument settings, and bioinformatic interpretation as one connected system.
FAQ
1. What is the most efficient method for protein sequencing?
For most modern research and analytical projects, LC-MS/MS is the most efficient method because it scales well, handles complex samples, and supports both identification and sequence interpretation.
2. Is Edman degradation still useful for protein sequencing?
Yes. It remains useful when a purified sample has an accessible N terminus and the main need is direct terminal confirmation rather than high-throughput proteome-scale analysis.
3. When should de novo sequencing be added?
Add de novo sequencing when databases are incomplete, when novel variants are expected, or when the project cannot rely solely on reference-driven peptide matching.
4. Can DNA sequencing replace protein sequencing?
No. DNA or RNA sequencing can predict an expected protein sequence, but it cannot directly confirm protein-level processing, PTMs, truncation, or sequence heterogeneity.
Conclusion
Efficient and accurate protein sequencing is really a workflow design problem. Mass spectrometry, de novo interpretation, Edman degradation, and terminal analysis each solve different parts of that problem. The most reliable results come from choosing the method that matches sample complexity, reference availability, and the exact level of sequence confirmation the study requires.
How to order?

