





Difference Between Re-Sequencing and De Novo Genome Assembly: How to Choose the Right Workflow for a New Genome Project
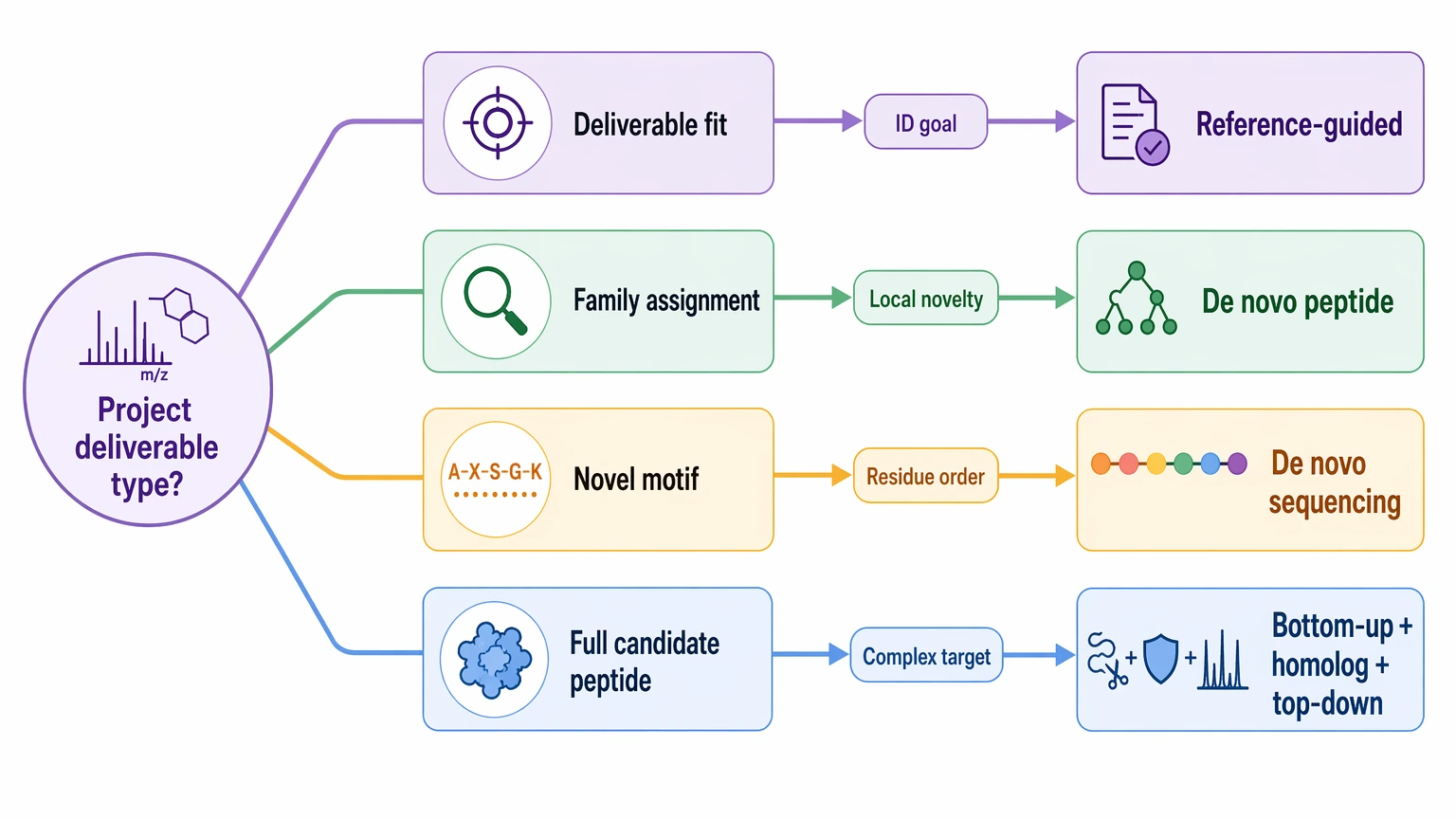
- Family assignment: reference-guided identification is often sufficient.
- Local novel motif discovery: de novo peptide sequencing may be enough even without full reconstruction.
- Candidate full peptide sequence proposal: de novo sequencing with overlapping peptide evidence is usually the better fit.
- Protein-level interpretation across a complex target: combine bottom-up proteomics with homolog comparison and, where feasible, top-down support.
- Publication, synthesis, or function-driven sequence decisions: plan orthogonal validation early.
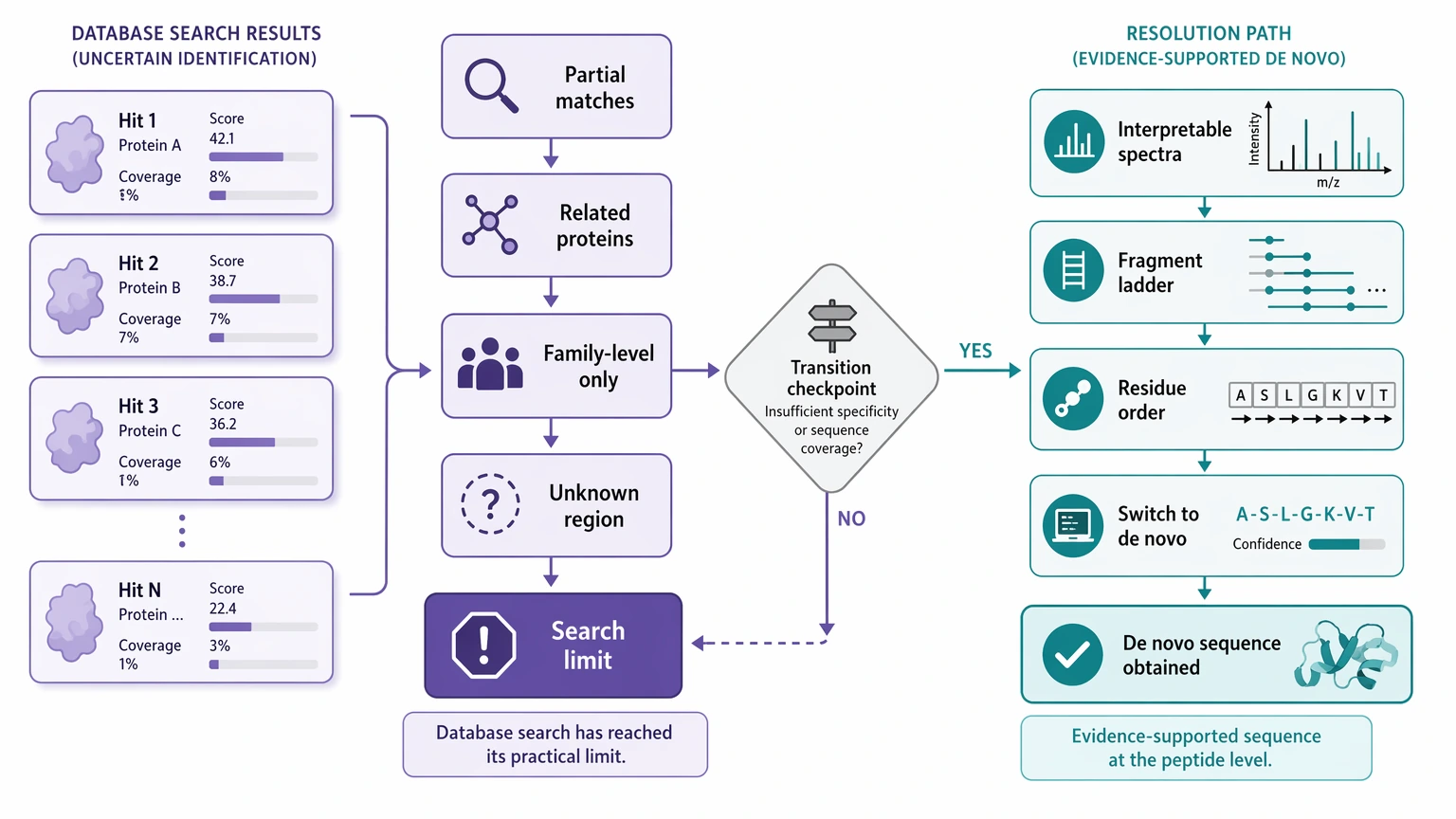
- repeated spectra do not support a confident PSM
- the best hits cluster around related proteins rather than one convincing target
- key peptides map only at the family level
- the biological question depends on the exact unknown region
Use reference-guided identification when your LC-MS/MS data line up well with a relevant database, show strong peptide-spectrum match (PSM) support, and the job is simply to confirm a known or closely related target. Shift to de novo peptide sequencing or de novo protein sequencing when the real sequence is absent from available databases, the sample is a non-reference sample, or the project needs a candidate sequence proposal instead of a family-level label.
Quick Workflow Decision
| Project signal | Better starting workflow | Why |
|---|---|---|
| Exact species or protein reference is available and matches multiple unique peptides | Reference-guided identification | Known sequence space can answer the question efficiently |
| Search results stop at homologous protein matches or conflicting IDs | Review search output, then shift toward de novo peptide sequencing | The database is no longer specific enough |
| Repeated MS/MS spectra remain unmatched in a non-reference sample | De novo peptide sequencing | Fragment-ion evidence may support direct sequence reconstruction |
| PTM-rich target on a known scaffold | Reference-guided identification first | The main question may be modification mapping, not sequence discovery |
| Exact novel sequence is required for synthesis or follow-up biology | De novo peptide sequencing or de novo protein sequencing | Identification alone does not resolve residue order |
A useful rule of thumb is simple: keep using database-backed identification while known sequence space still answers the biological question. Change course when the project needs sequence information the database cannot provide.
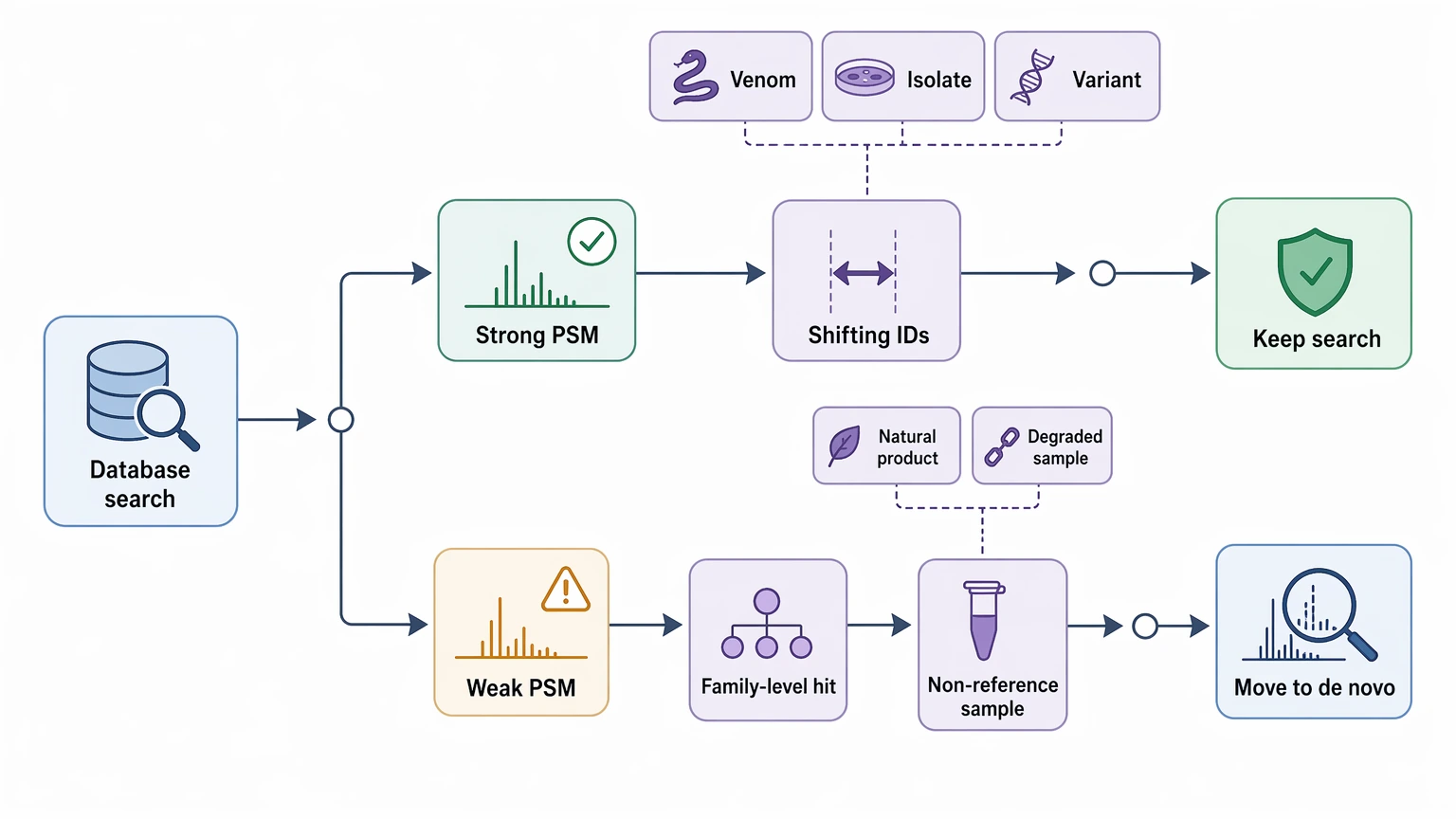
Where Teams Usually Reach This Decision
This choice usually comes up after an initial database search returns incomplete hits, weak PSM support, or assignments that shift from one related entry to another. That pattern shows up often in venom peptides, microbial isolates, engineered variants, natural products, and partially degraded samples.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
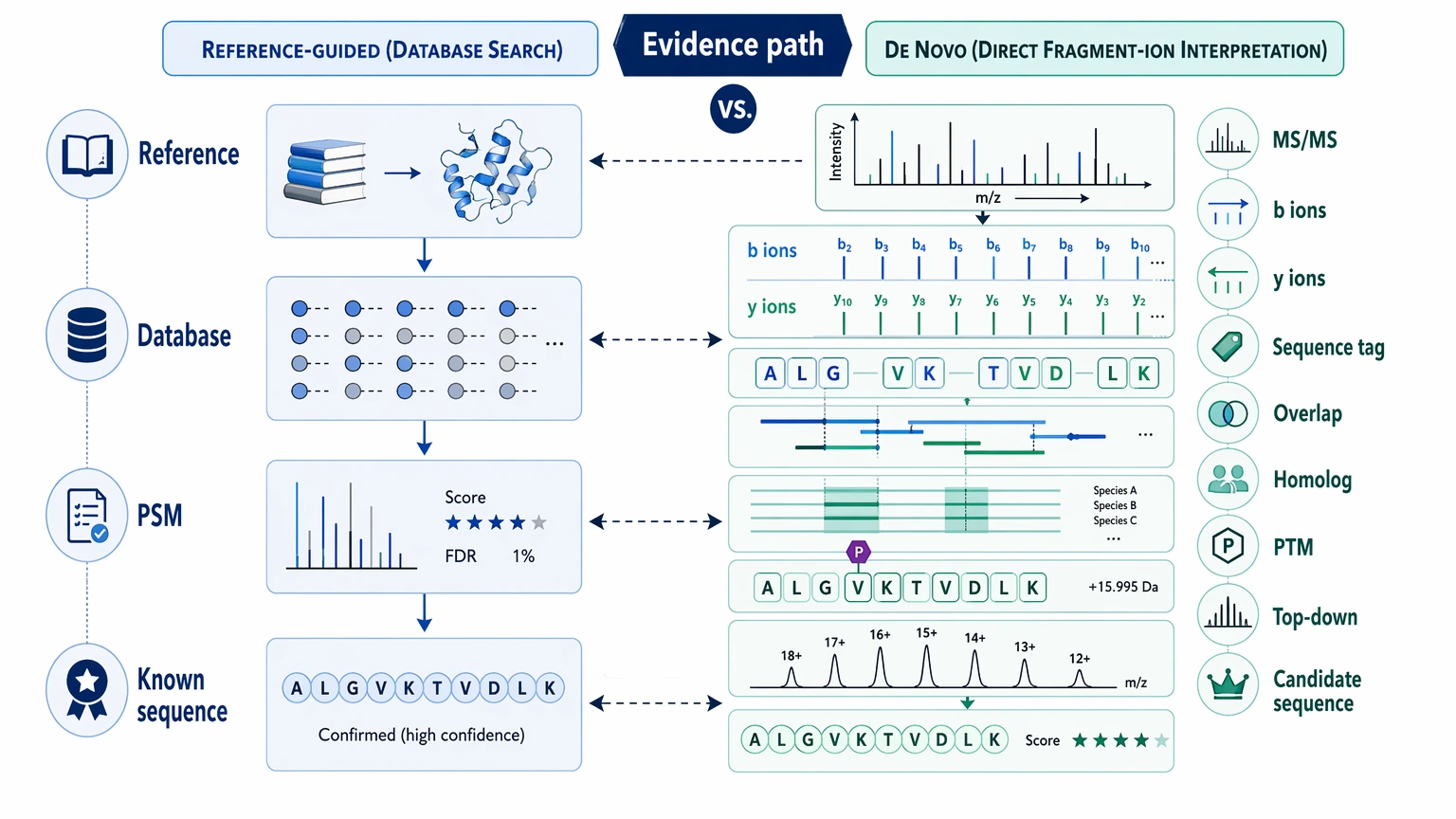
The distinction matters because the two workflows answer different questions. Reference-guided identification asks whether known sequence candidates can explain the spectra. De novo sequencing asks whether the tandem mass spectrometry (MS/MS) evidence itself can support residue order without depending on a correct database entry. If your question is still “what known protein family is this,” database search may be enough. If the question has changed to “what is the actual sequence,” you are now in de novo territory.
Practical Difference Between Reference-Guided Identification and De Novo Sequencing
Reference-guided identification follows a re-sequencing-style logic in LC-MS/MS. Spectra are matched against known sequence entries, and identity is inferred from the strongest-supported matches. This approach works well when the target, or a close homologous protein, is already represented in the database.
De novo peptide sequencing and de novo protein sequencing take a different path. They infer residue order directly from fragment ions, especially b ions / y ions, rather than assuming the right answer is already present in known sequence space. In practice, the workflow often begins with local sequence tag generation and then extends those tags through overlapping peptides, homolog comparison, PTM-aware interpretation, and sometimes intact protein / top-down support.
That difference changes the output. Reference-guided identification usually ends with a protein or family assignment plus sequence coverage statistics. De novo work produces a confidence-graded candidate sequence proposal, often with strong regions, weaker regions, and explicit residue ambiguity at certain positions. Even a strong de novo result is not the same as unconditional sequence truth.
Four Criteria That Drive the Workflow Choice
Reference availability and relevance
A large database is not automatically the right database. What matters is whether the exact target, or at least a biologically close sequence, is present and reasonably well annotated. If several unique peptides map cleanly to one reference and the biological question only requires identification, reference-guided identification is usually the faster and cleaner starting point.
The warning sign is not simply “few hits.” A stronger signal to switch is a pattern of partial matches across several related proteins, unstable ranking under different search settings, or a family-level assignment when the project really needs exact sequence confirmation.
MS/MS spectrum interpretability
De novo sequencing only works when the spectral evidence supports it. You need interpretable fragment-ion ladders, acceptable signal-to-noise behavior, and enough continuity in the ion series to support residue order across useful stretches of sequence.
If spectra are heavily mixed, sparse, or dominated by co-fragmentation, direct reconstruction becomes much less informative. In that situation, simplifying the sample or narrowing the question usually helps more than forcing a de novo pipeline.
Sequence novelty and PTM burden
Database search loses power when the target contains substitutions, truncation, fusion points, noncanonical segments, or a high post-translational modification (PTM) burden. Glycosylation, amidation, oxidation, cyclization, and disulfide-linked complexity can all interfere with routine matching.
That does not mean every modified sample belongs in a de novo workflow. If the sequence is already known and the project mainly needs PTM localization, reference-guided identification may still be the better first move. The real decision point is whether the unknown portion of the study is the modification pattern, the sequence itself, or both.
Deliverable fit
Projects often go off track because the expected output was never defined clearly enough. Some studies only need family-level identification. Others need a variable region, a set of high-confidence sequence tags, or a near-complete candidate peptide sequence.
Use the workflow that fits the actual deliverable:
When to Stop Forcing Database Search
Teams often stay in search mode longer than they should because partial matches keep appearing. Partial matches still have value, but they do not equal sequence resolution. A database-search route has probably reached its limit when the following pattern shows up together:
At that point, more searching may only confirm that the sample resembles something known. It may not show what is actually new. De novo peptide sequencing becomes easier to justify when the spectra contain enough interpretable fragment-ion evidence to support residue order in the region that matters.
Expected Results and How to Validate Them
Before you pick a workflow, define what “result” means for the project.
Immediate deliverables from reference-guided identification may include protein or family assignment, PSM evidence, peptide lists, and sequence coverage. Immediate deliverables from de novo work may include sequence tags, local high-confidence residue calls, PTM-aware interpretation, and a ranked candidate sequence proposal.
Follow-up confirmation is a separate step. If the sequence will guide synthesis, functional testing, or a single final claim, plan orthogonal validation such as targeted LC-MS/MS, synthetic peptide matching, additional digestion strategies, Edman-style confirmation where suitable, or top-down support for intact targets.
One limitation should always be stated plainly: MS/MS-based de novo interpretation can leave uncertainty at certain positions, especially with isobaric residues such as leucine and isoleucine, low-information regions, incomplete fragmentation, or PTM-rich spectra. Database-search limits and PTM complexity can also lower sequence confidence even when the overall interpretation still looks biologically plausible.
If you need help deciding whether your current raw files justify a de novo route, you can submit your requirements and LC-MS/MS context to MtoZ Biolabs to evaluate your project before you spend limited sample on follow-up confirmation.
Key Cautions and Practical Limits
No workflow removes all uncertainty, and several practical constraints affect whether de novo sequencing is realistic.
Sample quality or amount limits: low input, degradation, poor enrichment, or matrix interference can reduce usable spectra and shorten confident sequence tags.
Controls and repeat expectations: ambiguous assignments often need replicate acquisition, complementary digests, or targeted follow-up. One exploratory run may not be enough for a sequence-level decision.
Batch and contamination risk: mixed samples, carryover, co-isolation, and background contaminants can create misleading fragment patterns or inflate false protein inference.
Interpretation boundaries: a strong candidate sequence proposal is still evidence-weighted interpretation. It may support a publication or next experiment only after the key uncertain positions are tested directly.
When another route is better: if the project only needs family identification, known-target PTM mapping, or confirmation of one short variable region, a full de novo workflow may add work without changing the decision. Likewise, if spectra are too sparse or the sample is too heterogeneous, outside support for sample cleanup, acquisition redesign, or staged validation may be the better next step.
Choosing the Most Defensible Starting Point
The practical difference between re-sequencing-style identification and de novo reconstruction is not about which method sounds more advanced. It is about which question your evidence can answer. Reference-guided identification fits projects with relevant databases, coherent PSM support, and identification-level goals. De novo peptide sequencing or de novo protein sequencing fits projects where the database can no longer explain the biologically important region.
For a new LC-MS/MS sequence project, gather the sample source, purification status, expected PTMs, digest strategy, raw data availability, and desired deliverable before making the call. If the project involves a non-reference sample and the next decision depends on sequence-level evidence, contact MtoZ Biolabs to discuss the study, submit your requirements, and match the workflow to the spectra and validation burden you actually have.
FAQ
Can database search and de novo sequencing be combined in one project?
Yes. A common approach is to run database search first to capture known components, then apply de novo peptide sequencing only to the unresolved spectra or variable regions.
What kind of sample is the hardest for de novo sequencing?
Highly mixed samples are usually the hardest. Co-fragmentation and overlapping peptide signals make it much harder to assign a clean fragment-ion series to one sequence.
If I only need one variable region, do I still need full de novo protein sequencing?
Not always. A focused de novo peptide sequencing strategy for the informative region, followed by targeted confirmation, may answer the project question with less interpretation burden.
Can de novo sequencing help when the species is missing from public databases?
Often yes, as long as the MS/MS spectra are interpretable enough to support sequence tags and residue ordering. Poor spectral continuity can still limit what can be proposed with confidence.
What should I prepare before asking for workflow review?
Prepare raw LC-MS/MS files if available, sample origin, purification or enrichment details, digestion conditions, expected PTMs, and a clear statement of whether you need identification, a sequence tag, or a candidate sequence proposal.
How to order?

