





De Novo Sequencing Using Mass Spectrometry vs Database Search: When Is a Reference-Free Strategy Worth It?
- the reference database is species-matched or construct-matched
- the project needs routine identification rather than novel peptide identification
- homolog assignment is sufficient for the decision
- the MS/MS spectrum quality is mixed, but the reference space is strong
- the expected sequence is absent, weakly annotated, or biologically divergent
- the project needs direct evidence for a novel peptide, variant region, or undocumented construct segment
- the fragmentation spectrum contains interpretable fragment ions with useful b ions and y ions
- a sequence tag or locally confident sequence call would already move the project forward
- the sample contains both known and unexpected sequence content
- post-translational modification (PTM) complexity may distort ordinary peptide-spectrum match (PSM) scoring
- unmatched spectra, weak PSMs, or ambiguous regions need targeted reference-free review
- the species or construct is well represented in the reference database
- the goal is routine identification across many samples
- expected PTMs can be specified in a manageable search space
- throughput matters more than sequence novelty
- the needed answer is protein-level assignment rather than direct sequence reconstruction
- database search first, followed by de novo review of unmatched or weakly matched spectra
- de novo-derived sequence tag generation, then custom reference database refinement
- PTM-aware review around regions with unstable PSM support
- bottom-up proteomics evidence paired with targeted confirmation or limited top-down support
- targeted LC-MS/MS against candidate regions
- synthetic peptide comparison
- molecular biology sequence cross-checks for constructs or expressed products
- complementary digestion logic
- top-down support when peptide-level evidence is not enough for the protein-level conclusion
- Is the reference database truly relevant to the sample?
- Do you need a nearest known match, a sequence tag, or a defensible novel sequence claim?
- How clean are the MS/MS spectra, and how often do chimeric spectra appear?
- Are PTMs expected, heterogeneous, or unknown?
- Would a partial sequence already enable custom database refinement or targeted follow-up?
- How much ambiguity is acceptable in the final report?
A reference-free strategy is worth the added work when the sequence you need is likely absent, misleading, or too incomplete in the reference database to answer the real project question. When the reference database is relevant and the goal is routine protein inference rather than novelty, database search is usually the faster first option.
This article compares de novo sequencing using mass spectrometry with database search in the setting that matters most for project planning: limited-reference peptide or protein identification by LC-MS/MS. The real choice is not which method sounds more sophisticated. It is whether reference bias is a bigger risk than sequence ambiguity, interpretation workload, and the need for follow-up confirmation.
Quick Decision Block
Use database search first when:
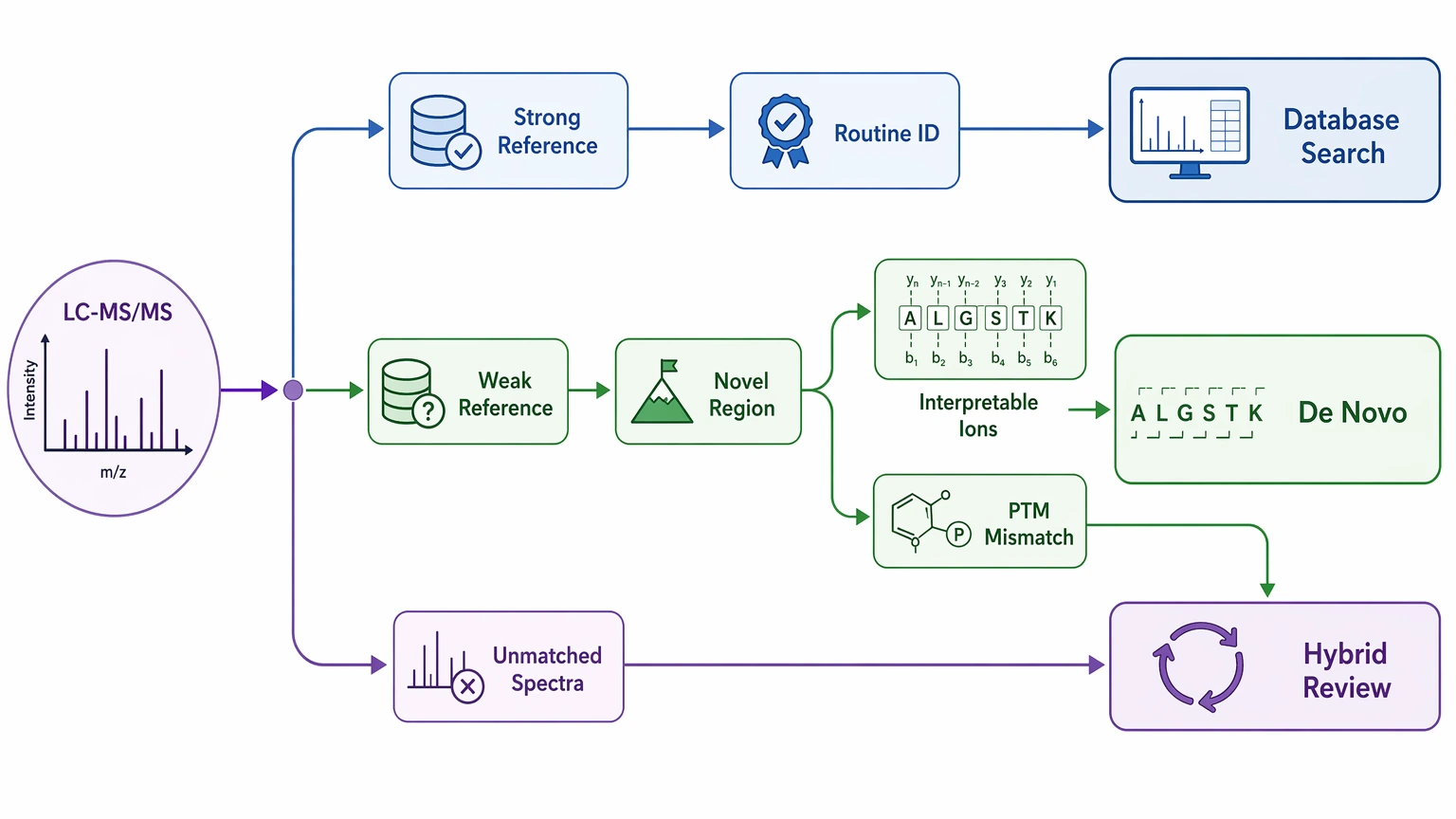
Use de novo sequencing using mass spectrometry when:
Use a hybrid workflow when:
What Each Workflow Actually Solves
A database search matches an observed MS/MS spectrum to theoretical fragment ions generated from a reference database. In bottom-up proteomics, this is often the quickest way to turn tandem mass spectrometry data into ranked PSMs and protein inference. Its main advantage is efficiency when the correct sequence is already in the search space.
De novo sequencing using mass spectrometry infers residue order directly from the fragmentation spectrum without requiring a complete reference database. That makes it useful when the true analyte is poorly represented, or not represented at all, in available references.
The practical difference is straightforward. Database search asks, “Which known entry best explains this spectrum?” De novo asks, “What sequence does this spectrum itself support?” Those questions overlap, but they stop being interchangeable when the reference space is weak.
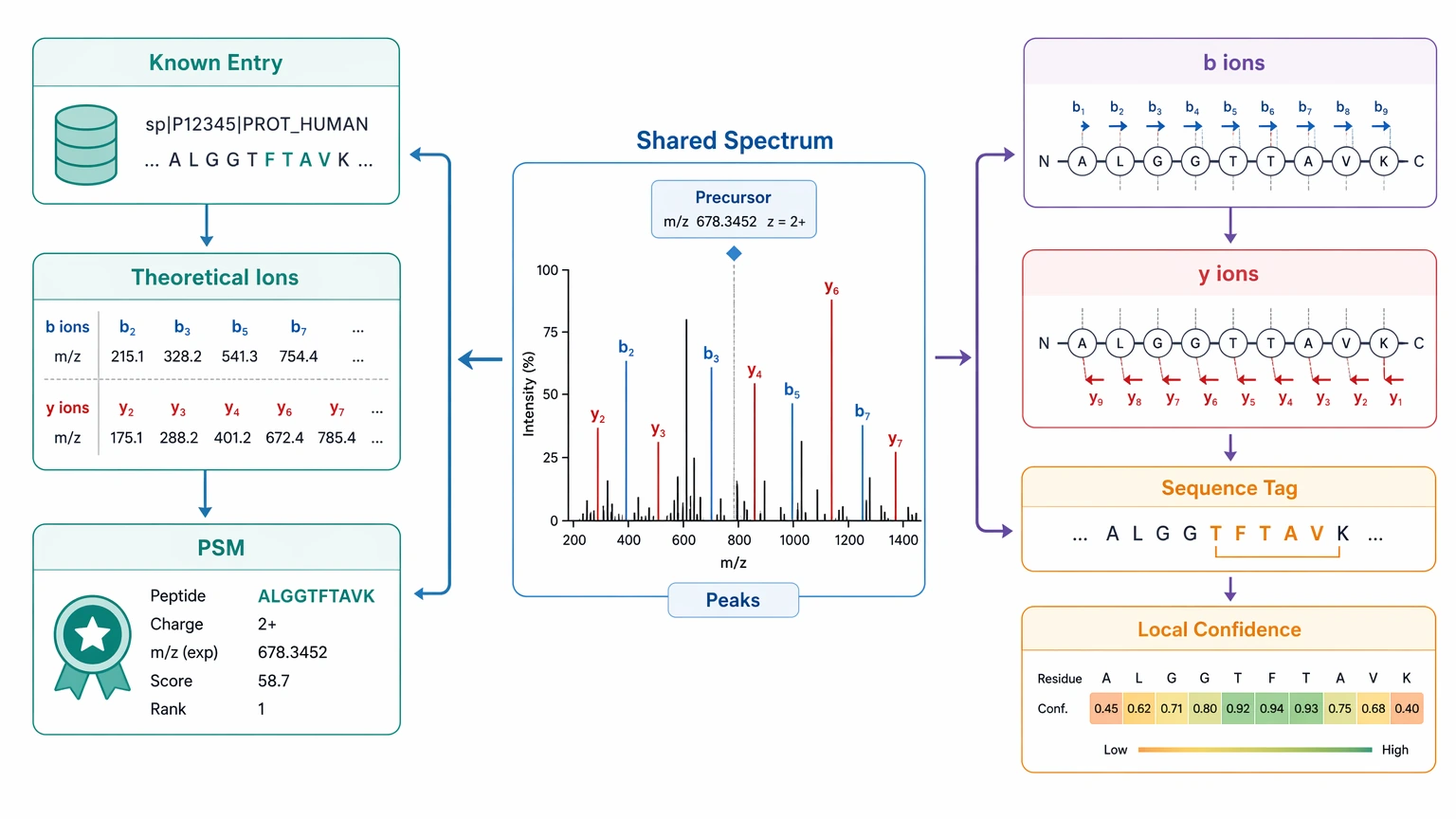
The Comparison Dimensions That Actually Drive the Decision
Four factors usually matter more than broad claims about software or workflow preference.
Reference database quality
If the reference database covers the true sequence space, database search is usually the better first-line choice. If it contains only distant or incomplete homologous entries, the top hit may still look persuasive while answering the wrong biological question.
MS/MS spectrum interpretability
A reference-free strategy only works when the fragmentation spectrum can support direct inference. Useful signs include clear precursor ion assignment, reasonable spectral quality, and enough fragment ions to support a defensible sequence tag or local sequence path.
Ambiguity burden
De novo outputs are not automatically complete or final. Isobaric residues, terminal uncertainty, incomplete sequence coverage, and local confidence gaps may still remain. The workflow makes sense only if those uncertainties are acceptable for the decision endpoint.
Validation burden
Routine database-linked identification is generally easier to report and defend. Novel or reference-free claims usually need more orthogonal validation before they are strong enough for publication, IP review, or a development decision.
Side-by-Side Comparison
The table below is most useful when your team is trying to decide whether the bigger risk comes from reference failure or from limited spectral interpretability.
| Dimension | Database search | De novo sequencing using mass spectrometry | Best fit |
|---|---|---|---|
| Reference dependence | Requires a relevant reference database | Works without a complete reference database | Weak or missing references favor de novo |
| Novel peptide identification | Constrained by existing entries | Better suited to absent or unexpected sequences | Novel sequence discovery |
| Spectral requirement | Can still work when reference support is strong | Needs interpretable fragment ions and sequence ladders | Cleaner MS/MS spectra |
| PTM handling | Strong for expected PTMs in a defined search space | Useful for mismatch review, but harder to interpret directly | PTM-rich or unexpected chemistry often needs hybrid review |
| Output type | PSMs, database-linked IDs, protein inference | Sequence tag, inferred peptide sequence, local confidence regions | Discovery or refinement work |
| Validation burden | Lower for known-sequence confirmation | Higher for novel claims | Orthogonal confirmation planned |
Takeaway: choose the workflow based on the dominant failure mode. If the main risk is a misleading reference database, add or prioritize de novo. If the main risk is weak fragmentation evidence, database-supported identification is often the safer route.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
When Database Search Is Usually Enough
Database search stays ahead in many common projects:
This is also the setting where false discovery rate (FDR) is easiest to apply and interpret, because FDR is defined within a database-driven matching framework. That advantage matters when projects need scalable reporting across large LC-MS/MS data sets.
When a Reference-Free Strategy Becomes Worth It
A reference-free strategy makes sense when database matching can give a false sense of confidence. Typical cases include non-model organisms, venom or natural peptide mixtures, engineered proteins with uncertain junctions, divergent homologs, and samples where the nearest known hit is not enough.
The central question is whether the project needs novel peptide identification or only nearest known assignment. If the decision depends on actual sequence content rather than the best available homologous entry, de novo becomes much more useful.
A second trigger is modification-related mismatch. Unexpected PTMs can shift fragment masses, reduce clean matching, and weaken otherwise solid PSM scoring. In that situation, de novo review can reveal sequence-supported regions that database search gives too little weight.
If you are comparing these routes for an active study, submit your requirements early so the planned deliverable matches the evidence level you actually need: homolog assignment, sequence tag, local sequence region, or a stronger novel sequence claim.
Hybrid Workflows Are Often the Most Defensible Option
For many projects, the best choice is neither database search alone nor de novo alone. A hybrid workflow can reduce reference bias without letting interpretation drift too far from the data.
Common hybrid designs include:
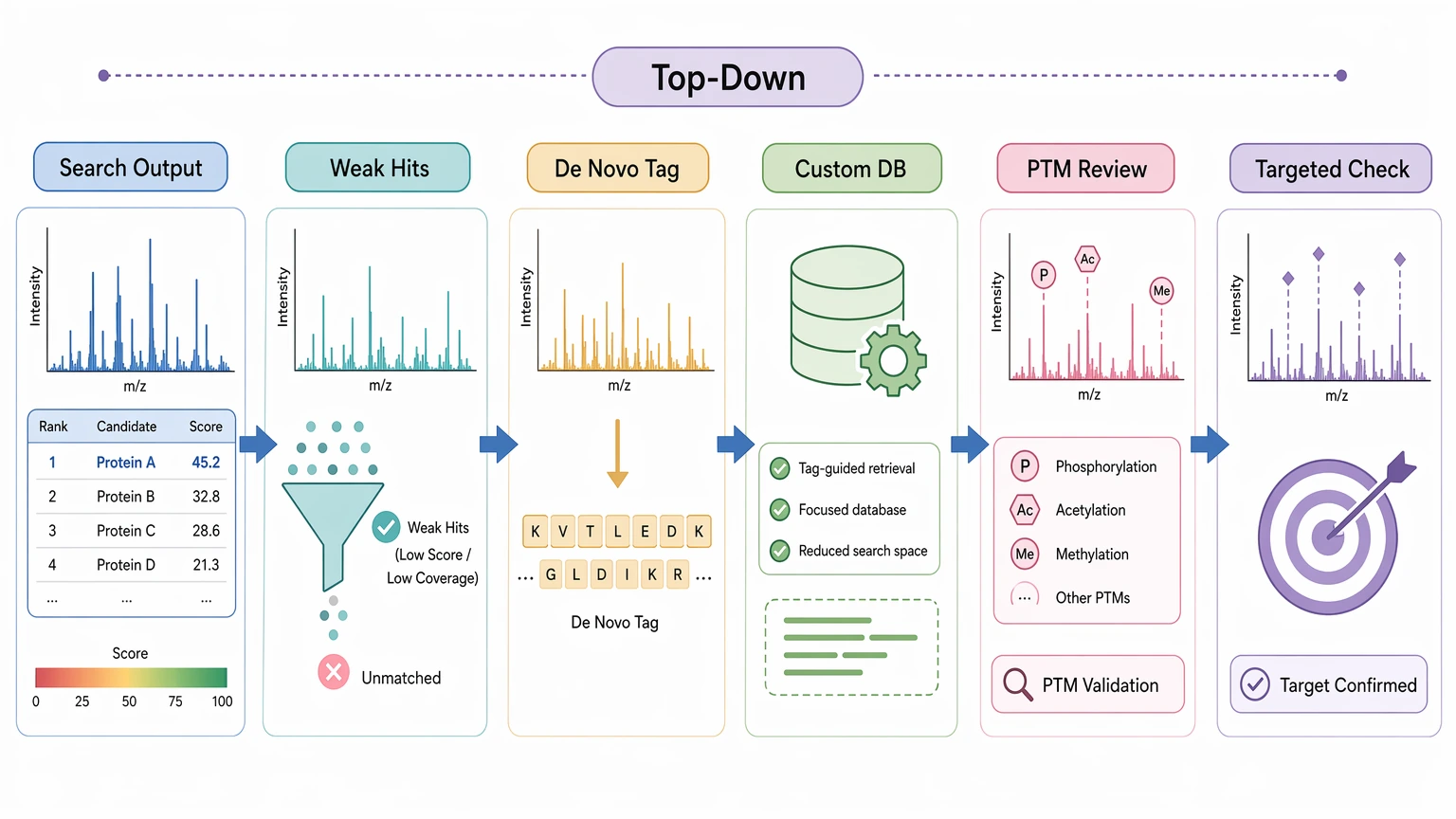
This approach is especially useful when only a subset of spectra truly challenges the reference model. You keep the efficiency of database search where it performs well and reserve reference-free interpretation for the spectra that matter most.
Expected Results and Validation Methods
Before choosing the workflow, decide what output will count as a usable result.
Immediate deliverables from database search often include ranked PSMs, peptide assignments, reference-linked proteins, and standard reporting fields. Immediate deliverables from de novo analysis are more likely to include a sequence tag, an inferred peptide sequence, confidence-limited local regions, or a shortlist of candidate interpretations.
Follow-up confirmation is a separate step. For novel sequence claims, useful orthogonal validation may include:
One limitation should be stated directly: de novo interpretation can remain uncertain even with good LC-MS/MS data, especially when PTMs alter fragment patterns, when a chimeric spectrum is present, or when sequence confidence depends on incomplete ion ladders rather than continuous backbone evidence.
Key Cautions and Practical Limits
A reference-free strategy becomes much less convincing when the underlying evidence is thin or mixed.
Sample quality or amount limits
Low abundance, impurity, degradation, or heavy matrix background can reduce spectral quality and shorten useful sequence coverage. Limited material also limits repeat acquisition and validation.
Controls and repeat expectations
Novel calls should not rest on a single borderline MS/MS spectrum when repeat data or a targeted follow-up experiment is feasible. Replicate support is not always mandatory, but it often changes confidence substantially.
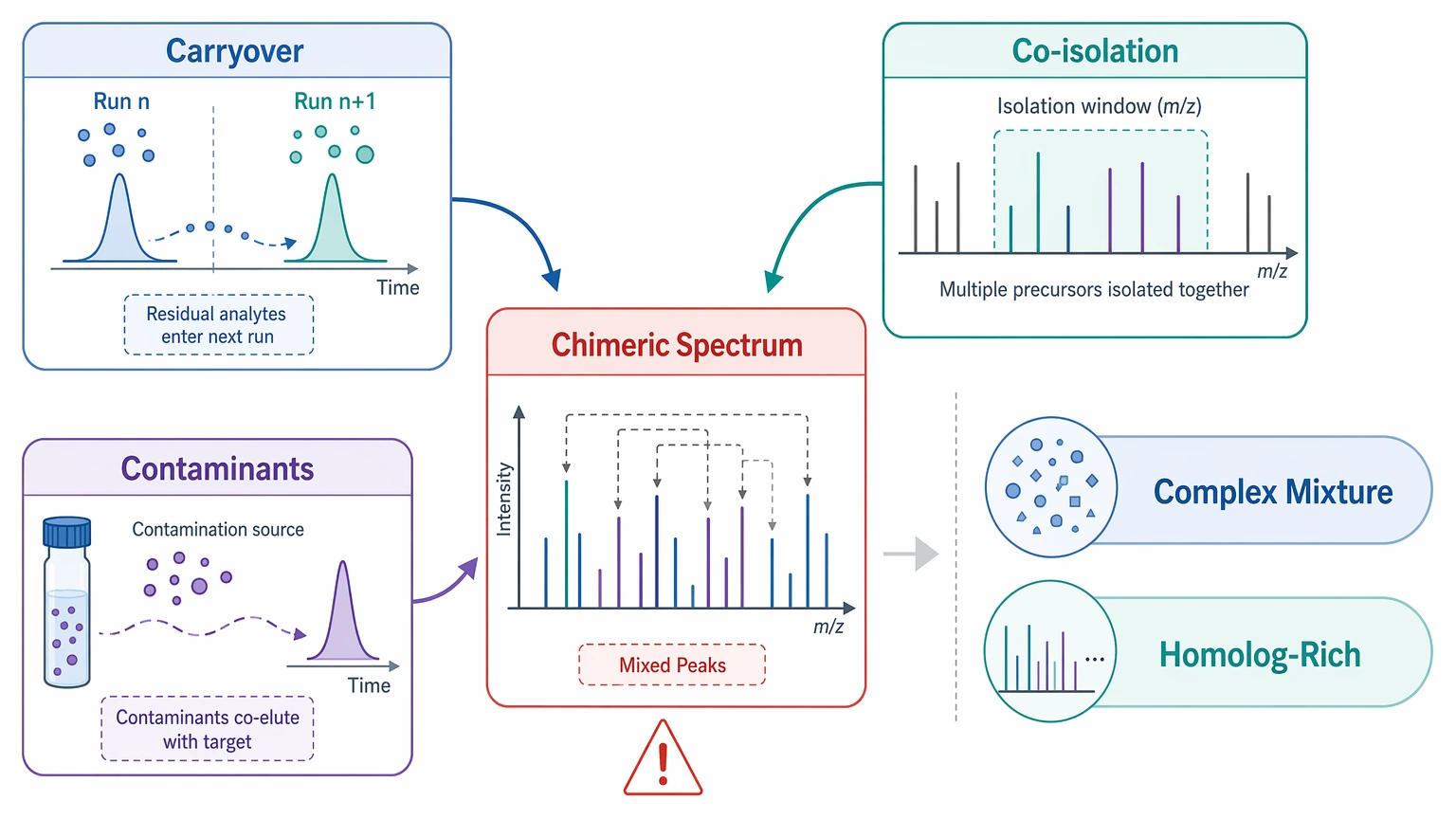
Batch effects and contamination risk
Carryover, co-isolation, and contaminant peptides can create a chimeric spectrum or distort fragment-ion interpretation. That risk matters most in complex mixtures and homolog-rich samples.
Interpretation boundaries
Leucine/isoleucine ambiguity remains a classic example of unresolved isobaric residues in standard tandem mass spectrometry. PTM localization may also stay local rather than definitive if fragment ions do not bracket the modified site. Peptide-level evidence should not be overstated as full protein sequencing without clear assembly logic.
When another method is the better next step
If the real need is known-sequence confirmation, peptide mapping or targeted validation may be more efficient than a de novo-first plan. If protein-level architecture matters more than partial peptide inference, top-down support or sequence cross-checking outside LC-MS/MS may be the better next move.
Project Readiness Questions Before You Commit
A workflow decision gets easier when the team can answer a few practical questions:
If your team needs help sorting through those trade-offs, MtoZ Biolabs can evaluate the sample context, LC-MS/MS evidence, and validation path so you can submit your requirements with a workflow that fits the decision you actually need to make.
Comparison Summary and Next Step
Database search is usually the right first move when the reference database is strong and the project needs efficient identification of expected sequences. De novo sequencing using mass spectrometry becomes worth the added interpretation burden when reference bias is likely to hide the sequence feature that matters most. Between those two ends, hybrid workflows often give the most decision-useful answer.
For non-model organisms, engineered constructs, PTM-rich analytes, and novel peptide discovery projects, the best plan depends on the evidence you will ultimately need to defend and the amount of ambiguity your project can tolerate. If you are preparing a sequencing study, contact MtoZ Biolabs to evaluate your project, discuss the sample and LC-MS/MS context, and align the workflow with the level of validation the final claim will require.
FAQ
Can a strong database search still miss the biologically important answer?
Yes. A high-scoring PSM can still point to the nearest known entry rather than the true analyte if the reference database lacks the sequence feature that matters for the project, such as a variant junction, divergent homolog, or unexpected truncation.
Is de novo sequencing useful if it only produces a sequence tag?
Often yes. A sequence tag can be enough to refine a custom reference database, narrow a candidate list, design targeted follow-up, or test whether a claimed novel region is plausible.
Why is FDR harder to compare between database search and de novo outputs?
FDR is built around database-driven match competition. De novo confidence depends more directly on fragment-ion evidence, ambiguity structure, and the strength of follow-up confirmation, so the numbers are not directly interchangeable.
Are peptide-level de novo results the same as protein-level sequencing?
No. Peptide-level inference may support only part of a protein claim. Full protein-level conclusions can require assembly across peptides, complementary digestion, or another confirmation layer.
What sample types most often justify a hybrid workflow?
Samples with partial reference coverage, mixed known and unknown content, or PTM-related mismatch are strong candidates. Engineered proteins, venom peptides, and weakly annotated species are common examples.
What should be prepared before asking for a workflow assessment?
Bring the sample type, purification status, organism or construct context, available LC-MS/MS data, expected PTMs, and the confidence level needed for the final decision or report.
How to order?

